Copyright2000 by the Genetics Society of America
Functional Interaction Between the
PKC1
Pathway and
CDC31
Network of SPB Duplication Genes
Waheeda Khalfan,
1Irena Ivanovska
1and Mark D. Rose
Department of Molecular Biology, Princeton University, Princeton, New Jersey 08544-1014
Manuscript received February 25, 2000 Accepted for publication April 12, 2000
ABSTRACT
The earliest known step in yeast spindle pole body (SPB) duplication requires Cdc31p and Kar1p, two physically interacting SPB components, and Dsk2p and Rad23p, a pair of ubiquitin-like proteins. Compo-nents of the PKC1 pathway were found to interact with these SPB duplication genes in two independent genetic screens. Initially, SLG1 and PKC1 were obtained as high-copy suppressors of dsk2⌬rad23⌬and a mutation in MPK1 was synthetically lethal with kar1-⌬17. Subsequently, we demonstrated extensive genetic
interactions between the PKC1 pathway and the SPB duplication mutants that affect Cdc31p function. The genetic interactions are unlikely to be related to the cell-wall integrity function of the PKC1 pathway because the SPB mutants did not exhibit cell-wall defects. Overexpression of multiple PKC1 pathway components suppressed the G2/M arrest of the SPB duplication mutants and mutations in MPK1 exacer-bated the cell cycle arrest of kar1-⌬17, suggesting a role for the PKC1 pathway in SPB duplication. We
also found that mutations in SPC110, which encodes a major SPB component, showed genetic interactions with both CDC31 and the PKC1 pathway. In support of the model that the PKC1 pathway regulates SPB duplication, one of the phosphorylated forms of Spc110p was absent in pkc1 and mpk1⌬mutants.
S
ACCHAROMYCES cerevisiae cells commit to a new Treisman 1997;Watanabe et al. 1997) that regulates round of cell division during G1. Activation of the transcription of a number of cell-wall components G1-specific Cdc28p/cyclin complex promotes bud (JungandLevin 1999). In addition, Mpk1p may also emergence, DNA replication, and duplication of the target the transcription factor complex Swi4p/Swi6p yeast microtubule organizing center, the spindle pole (Maddenet al. 1997).body (SPB;PringleandHartwell1981;Reed1992; Although duplication of the SPB in G1 is also Cdc28p/ Cross1995). These major G1 cell cycle events occur Cln dependent, the specific signals that activate SPB independently of each other, but in a coordinated man- duplication are unknown. The yeast SPB is a trilaminar ner that has yet to be understood. disc-like structure embedded in the nuclear envelope The PKC1 cell integrity pathway has been implicated from which nuclear and cytoplasmic microtubules are in numerous cellular processes including promoting organized (ByersandGoetsch1974, 1975). The earli-bud emergence at the G1/S transition (Mazzoniet al. est step in SPB duplication involves the formation of an 1993;Marini et al. 1996; Zarzov et al. 1996; Gray et electron-dense material on the cytoplasmic face of the al. 1997;Igual et al. 1997). Pkc1p (Levin et al. 1990; SPB, called the satellite. The satellite is the precursor Paraviciniet al. 1992;Yoshidaet al. 1992) activates a of the nascent SPB, or the spindle plaque, which forms mitogen-activated protein (MAP) kinase cascade con- in the cytoplasm and is then embedded into the nuclear sisting of Bck1p (LeeandLevin1992), Mkk1p/Mkk2p envelope (Adams and Kilmartin 1999). The early (Irieet al. 1993), and the MAP kinase, Mpk1p (Leeet stages of SPB duplication are independent of Cdc28p al. 1993). The PKC1 pathway is most likely regulated by activity since mutations in CDC28 result in cell cycle the small GTP-binding protein Rho1p in vivo (Nonaka arrest with a single SPB already containing a satellite et al. 1995;Kamadaet al. 1996). Slg1p/Wsc1p, a plasma (ByersandGoetsch1974). This observation suggests membrane protein, acts through Rho1p to activate the
that the signal to initiate SPB duplication is active before PKC1 pathway (Gray et al. 1997; Verna et al. 1997;
the Cdc28p/START point, but for SPB duplication to Jacobyet al. 1998). The MAP kinase Mpk1p
phosphory-continue, a CDC28-dependent signal is required. lates downstream targets including Rlm1p, a MADS-box
Analysis of the early SPB duplication genes may give transcription factor (Watanabeet al. 1995;Dodouand
insight into how cell cycle control is exerted on SPB duplication because the G1 cell cycle machinery would be expected to target proteins required in the early
Corresponding author: Mark D. Rose, Department of Molecular
Biol-stages of SPB duplication. Mutations in CDC31, the yeast
ogy, Princeton University, Princeton, NJ 08544-1014.
homolog of centrin, block the earliest stage of SPB
du-E-mail: [email protected]
1These authors contributed equally to this work. plication, the formation of the satellite precursor
(Byers1981;Schildet al. 1981;Baumet al. 1986). In possibly to coordinate SPB duplication and bud emer-gence during G1.
addition to SPB duplication, Cdc31p also plays a role in morphogenesis and cell integrity via interaction with Kic1p (Sullivan et al. 1998). Mutations in the kinase
MATERIALS AND METHODS domain of Kic1p and certain cdc31 alleles result in
ab-normal bud morphology and lysis. However, kic1 and Microbial techniques and yeast strain construction: Yeast media and microbial techniques were essentially as previously cdc31 mutants exhibit cell-wall defects that appear to be
described (Roseet al. 1990). Bacterial media were as described different than those observed in pkc1 pathway mutants
(Sambrook et al. 1989), and bacterial strain XL1-Blue was (Sullivanet al. 1998). used for all bacterial manipulations. All bacterial strains are
Another SPB component, Kar1p, is required at the listed in Table 1.
All yeast strains used are listed in Table 2. Yeast strains same step of SPB assembly as Cdc31p (RoseandFink
were constructed using standard genetic techniques. All yeast 1987). Kar1p genetically and physically interacts with
strains were isogenic to S288c. To generate double mutants Cdc31p and is required to localize Cdc31p to the SPB
for synthetic lethality studies, strains with mutations in cdc31-1, (BigginsandRose1994;Vallenet al. 1994;Spanget kar1-⌬17, spc10-220, mps2-1, and dsk2⌬rad23⌬were kept cov-al. 1995). The temperature-sensitive mutation kar1-⌬17 ered with a URA3 plasmid bearing the appropriate wild-type gene to prevent increased ploidy. PKC1 pathway components mislocalizes Cdc31p at high temperature and can be
were then disrupted in the covered mutant strains. The re-rescued by high dosage of CDC31 and the dominant
sulting double mutant strains containing wild-type (WT) URA3 allele CDC31-16. Formally, therefore, Kar1p functions plasmids were streaked on 5-fluoroorotic acid (5-FOA) at 23 upstream of Cdc31p. DSK2 is a nonessential ubiquitin- to select for plasmid loss (Boekeet al. 1984). When double like protein identified as a dominant suppressor that or triple mutant combinations were viable on 5-FOA at 23⬚, growth at higher temperatures was assayed by spotting 10-fold also relocalizes Cdc31p in kar1-⌬17 mutants (Biggins
serial dilutions. andRose1994;Vallenet al. 1994;Bigginset al. 1996).
In most cases, double mutants between PKC1 pathway com-Deletion of DSK2 in combination with deletion of an- ponents and SPB duplication genes were generated either by other ubiquitin-like gene, RAD23, results in a tempera- PCR-mediated gene disruption (Baudin et al. 1993) or with
disruption plasmids listed in Table 1. In a few cases, the dou-ture-sensitive block in SPB duplication prior to satellite
ble/triple mutants were generated as segregants of genetic formation (Biggins et al. 1996). Unlike mutations in
crosses. Specifically, the slg1⌬kar1-⌬17 strain (MY4646) was KAR1, the dsk2⌬ rad23⌬ strain is not defective for constructed by mating a kar1-⌬
17 [KAR1 URA3] (MS2373) Cdc31p localization at the SPB. However, like kar1-⌬17 strain to a slg1⌬ (MY4455), a cdc31-1 slg1⌬ double mutant mutants, the SPB duplication defect of dsk2⌬ rad23⌬ (MY6862) by crossing MY4455 to MY3885, the dsk2⌬rad23⌬ slg1⌬(MY4916) strain by mating MY4449 to MY4455, the pkc1ts
can be suppressed by high-copy CDC31 and by
CDC31-kar1-⌬17 strain (MY6805) by crossing DL523 (Levin and 16. These results suggest that Dsk2p/Rad23p also
func-Bartlett-Heubusch1992) to MS2374, and a dsk2⌬rad23⌬ tion upstream of Cdc31p to mediate an important func- pkc1ts strain (MY6296) by mating DL523 (Levin and
Bart-tion of Cdc31p at the SPB (Bigginset al. 1996). lett-Heubusch1992) to MY4197.
kar1-⌬17 synthetic lethality screen and identification of By employing the SPB duplication mutants kar1-⌬17
mpk1:To identify mutations that result in synthetic lethality and dsk2⌬rad23⌬in different genetic screens, we
identi-with kar1-⌬17, a “shuffle-mutagenesis” screening strategy was fied multiple genetic interactions between the PKC1 and employed. MS2373 and MS2376, ura3 kar1-⌬
17 strains of both the SPB duplication pathways. Members of the PKC1 mating types, were constructed, each carrying a KAR1 URA3-pathway, in high copy, could suppress the temperature based centromeric plasmid to suppress the mutant defect. The strains were mutagenized with ethyl methanesulfonate (Rose
sensitivity of kar1-⌬17, dsk2⌬rad23⌬, and cdc31-2.
Over-et al. 1990) to 40% viability and plated on synthOver-etic complOver-ete expression of different members of the PKC1 pathway
medium lacking uracil (SC-ura) to select for the covering specifically suppressed the SPB duplication mutants but plasmid. Mutations that cause slow growth or lethality after not other G2/M-arrested cells. In addition, we analyzed loss of the covering plasmid were assayed by their sensitivity to 5-FOA media, which selects against the URA3 plasmid. Out many combinations of double mutants and found
exten-of 60,000 colonies screened, we identified four 5-FOA-sensitive sive synthetic lethality and synthetic growth defects. To
mutants whose 5-FOA sensitivity was recessive and segregated understand the functional basis of the synthetic interac- as single genes. The 5-FOA sensitivity of the four mutants was tions, we analyzed the phenotypes of the double mutants rescued by a second plasmid bearing KAR1 on a LEU2-based with respect to cell integrity and G2/M arrest. In a vector, as expected for mutations that affect KAR1 function. Complementation analysis revealed that two of the mutations mpk1⌬kar1-⌬17 double mutant, the G2/M arrest defect
defined unique linkage groups and were subsequently identi-was clearly exacerbated. We also found that
phosphory-fied as mutations in REG1 and NEM1 (W. Khalfanand M. D. lation of Spc110p, an essential SPB component, was Rose, unpublished observations). The remaining two muta-defective in pkc1tsand mpk1⌬mutants, suggesting that
tions, SL6 and SL18, belonged to the same linkage group and caused temperature sensitivity at 37⬚, and the temperature the PKC1 pathway may directly or indirectly regulate
sensitivity was linked to 5-FOA sensitivity because they cose-Spc110p phosphorylation. Our results demonstrate an
gregated in crosses (⬍3.1 cM).
TABLE 1
Bacterial Plasmids
Plasmid Yeast marker Source
p10 PKC1 URA3 2 D. Levin (Johns Hopkins University)
p636 BCK1-20 URA3 CEN D. Levin
p666 MPK1 LEU2 CEN D. Levin
p759 mpk1⌬::TRP D. Levin
p784 bck1⌬::HIS3 D. Levin
pBG168 CDC13 URA3 CEN L. Hartwell (Fred Hutchinson Cancer Research Center) pHS40 spc110-220 URA3 YIP T. Davis (University of Washington, Seattle)
pJG109 SPC110 URA3 CEN T. Davis
pMR2012 CDC31 URA3 2 Vallenet al. (1994)
pMR2223 CDC31-16 URA3 CEN ARS Vallenet al. (1994)
pMR2745 DSK2-1 URA3 CEN ARS Bigginset al. (1996)
pMR2757 DSK2 URA3 CEN ARS Bigginset al. (1996)
pMR4090 SLG1 URA3 2 This study pMR4290 MPK1 URA3 2 This study
pMR4481 MPS2 URA3 CEN Mark Winey (University of Colorado, Boulder) pMR76 KAR1 URA3 CEN ARS RoseandFink(1987)
pMR77 KAR1 URA3 CEN ARS RoseandFink(1987) pRH102 CDC7 URA3 CEN Hollingsworthet al. (1992)
pRS416 URA3 CEN SikorskiandHieter(1989) pRS426 URA3 2 SikorskiandHieter(1989)
pSJ0 CDC15 URA3 CEN D. O. Morgan (University of California, San Francisco)
(MS5589) strain. MS5589 was transformed with a URA3 further. To identify suppressors that were specific for SPB duplication, the plasmids were tested for their ability to sup-YCp50 library (Rose et al. 1987). Of 18,000 transformants
examined, 7 were Ts⫹. All seven plasmids rescued the tempera- press KAR1 and CDC31 mutations. Nine plasmids partially suppressed CDC31 mutations in an allele-specific manner. To ture sensitivity of SL6 when reintroduced into yeast. The insert
junctions of three plasmids were sequenced and contained identify the suppressing genes, we sequenced the ends of the library inserts using primers from the YEp24 vector. Two of two open reading frames in common, YHR029C and MPK1/
SLT2. A plasmid containing only the MPK1/SLT2 gene (p666, them contained the SLG1 gene and were strong suppressors of both kar1-⌬17 and cdc31. One plasmid contained the PKC1
kindly provided by Dr. Levin), rescued the temperature
sensi-tivity of MS5589 and the 5-FOA sensisensi-tivity of MS5584. To gene.
Microscopy:To examine the G2/M arrest phenotype of the confirm that the MPK1 gene and SL6 locus were allelic, an
mpk1⌬ kar1-⌬17 double mutant (MS5886) was generated, SPB duplication mutants, strains were grown at 23⬚ to early logarithmic phase and one-half of the cultures were shifted shown to be 5-FOA sensitive, and the 5-FOA sensitivity could be
suppressed with a LEU2 MPK1 plasmid. To establish linkage, to the indicated temperatures for 4-8 hr. To examine the nuclear morphology, 1 ml of each culture was collected by MS5831, a strain containing the SL6 mutation alone, was
crossed to a mpk1⌬strain (MS5933). The resulting diploid centrifugation and stained with 4⬘,6-diamidino-2-phenylindole (DAPI) essentially as described (Roseet al. 1990). DAPI was
strain was Ts⫺, indicating that mpk1⌬and SL6 fail to
comple-ment. Because a homozygous mpk1⌬ diploid cannot sporu- obtained from Accurate Biochemicals and Scientific Corp. (Westbury, NY).
late, an MPK1 LEU2 plasmid was introduced into the diploid.
Upon sporulation, all spores that did not contain the MPK1 For cell cycle analysis of mpk1⌬kar1-⌬17, strain MS5886 was
grown on 5-FOA at 23⬚for several days until small,
slow-grow-LEU2 plasmid (43/43) were temperature sensitive, indicating
that there were no wild-type recombinants. We concluded ing colonies appeared (incubation at lower temperatures failed to give any colonies). As a control, strain MS2373 was that MPK1 and SL6 are allelic because they are closely linked
(⬍4.7 cM). also grown on 5-FOA at 23⬚. The colonies arising on the 5-FOA plate were propagated in YPD liquid where the double
dsk2⌬rad23⌬high-copy suppressor screen: Strain MY3592
was transformed with a YEp24 genomic library (Carlsonand mutant strain continued to grow very slowly compared to the control strain.
Botstein1982). A total of 21,300 Ura⫹ transformants (17
genome equivalents) were selected on SC-ura medium at 30⬚ For live/dead analysis, strains were grown as described above. FUN-1 dye (Millardet al. 1997), was added to 1 ml
and subsequently replica printed to SC-ura at 37⬚. Thirty
puta-tive Ts⫹ colonies were picked and retested. Plasmids were of culture to a final concentration of 10m(Molecular Probes, Eugene, OR). The cultures were incubated at room tempera-recovered from yeast as previously described (Roseet al. 1990)
and plasmid linkage of the Ts⫹ phenotype was tested after ture, in the dark, for 0.5 hr. Cells were examined by differential interference and fluorescence microscopy (Axiophot, Carl retransformation into MY3592. The major class of plasmids
(11) contained the CDC31 gene. RAD23 was isolated twice. Zeiss, Inc., Thornwood, NY).
Spc110 Western blot analysis: Affinity-purified Spc110
anti-DSK2 was not isolated, presumably because of its toxicity when
overexpressed. Two independent secondary screens were used bodies were obtained from T. Davis (University of Washington, Seattle, WA) and crude Spc110 antibodies were obtained from to classify the remaining 17 suppressors. Because rad23⌬, but
TABLE 2
Saccharomyces cerevisiaestrains
Strain Genotype Source
2032 MATa ura3 omnc cdc7-1 TRP⫹ a
DL523 MATa ura3-52 trp1-1 his4 can1Rpkc1⌬::LEU2 [pkc1tsYcp50] b
HSY11-4D MATa ura3-1 ade2-1oc can1-100 leu2-3, 112 trp1-1 ade3⌬spc110-220 c
MS1554 MATa ura3-52 leu2-3, 112 ade2-101 his3-⌬200
MS2083 MATa ura3-52 leu2-3, 112 trp1⌬1 kar1-⌬17
MS2374 MAT␣ura3-52 leu2-3, 112 trp1⌬1 ade2-101 kar1-⌬17
MS2373 MATa ura3-52 leu2-3, 112 trp1⌬1 kar1⌬17 [MR76]
MS2376 MAT␣ura3-52 leu2-3, 112 ade2-101 kar1⌬17 [MR76]
MS5584 MAT␣ura3-52 leu2-3, 112 ade2-101 kar1⌬17 mpk1 (SL6) [MR76]
MS5589 MAT␣ura3-52 leu2-3, 112 ade2-101 kar1⌬17 mpk1 (SL6) [MR326]
MS5820 MAT␣ura3-52 leu2-3, 112 ade2-101 his3-⌬200 kar1⌬17 [MR76]
MS5831 MAT␣ura3-52 leu2-3, 112 ade2-101 mpk1 (SL6)
MS5886 MATa ura3-52 leu2-3, 112 trp1⌬1 kar1⌬17 mpk1⌬::TRP1 [MR76]
MS5888 MAT␣ura3-52 leu2-3, 112 ade2-101 hsi3-⌬200 kar1⌬17 bck1⌬::HIS3 [MR76]
MS5930 MATa ura3-52 leu2-3, 112 trp1⌬1 mpk1⌬::TRP1
MS5933 MATa ura3-52 leu2-3, 112 mpk1⌬::TRP1
MS5938 MAT␣ura3-52 leu2-3, 112 ade2-101 his3-⌬200 bck1⌬::HIS3
MS6547 MATa ura3-52 leu2-3, 112 ade2-101 his3-⌬200 mpk1⌬::HIS3
MS6934 MATa ura3-52 ade2-101 his3-⌬200 leu2-3, 112 swi4⌬::HIS3
MS6937 MATa ura3-52 ade2-101 his3-⌬200 leu2-3, 112 rlml⌬::HIS3
MS6944 MAT␣ura3-52 ade2-101 his3-⌬200 leu2-3, 112 swi4⌬::HIS3 kar1⌬17 [MR76]
MS6947 MAT␣ura3-52 ade2-101 his3-⌬200 leu2-3, 112 rlm1⌬::HIS3 kar1⌬17 [MR76]
MY2259 MAT␣ura3 leu2-3, 112 his7 trp1 cdc31-1 d
MY2260 MAT␣ura3 leu2-3, 112 his7 trp1 ade2 cyh2Rcdc31-2 d
MY3592 MAT␣ura3-52 leu2 ade2 his3 lys2 dsk2⌬::LEU2 rad23⌬
MY3752 MATa ura3-52 leu2-3, 112 ade2-101 kar1-⌬17 [MR2223]
MY3753 MATa ura3-52 leu2-3, 112 ade2-101 kar1-⌬17 [MR2032]
MY3754 MATa ura3-52 leu2-3, 112 ade2-101 kar1-⌬17 [MR2745]
MY3755 MATa ura3-52 leu2-3, 112 ade-101 kar1-⌬17 [pRS416]
MY3883 MATa ura3-52 ade2-101 leu2-3, 112 his3-⌬200 cdc31-1 [MR2012]
MY3885 MATa ura3-52 ade2-101 leu2-3, 112 cdc31-1
MY4193 MAT␣ura3-52 leu2 ade2 his3-⌬200 dsk2⌬::LEU2 rad23⌬
MY4197 MAT␣ura3-52 ade2 leu2 his3-⌬200 dsk2⌬::LEU2 rad23⌬::URA3
MY4449 MATa ura3-52 leu2 ade2 his3-⌬200 dsk⌬::LEU2 rad23⌬::URA3
MY4455 MAT␣ura3-52 leu2 his3-⌬200 trp1-⌬63 lys2-801 slg1⌬::LEU2
MY4642 MAT␣ura3-52 leu2-3, 112 his3-⌬200 kar1-⌬17 slg1⌬::LEU2
MY4646 MATa ura3-52 leu2-3, 112 his3-⌬200 kar1-⌬17 slg1⌬::LEU2
MY4916 MAT␣ura3-52 leu2-3, 112 lys1-801 ade2 his3-⌬200 dsk2⌬::LEU2 rad23⌬::URA3 slg1⌬::LEU2
MY5064 MAT␣ura3-52 leu2 ade2-101 his3-⌬200 dsk2⌬::LEU2 rad23⌬[MR2757] MY5689 MAT␣ura3-52 leu2-3, 112 his3-⌬200 dsk2⌬::LEU2 rad23⌬slg1⌬::LEU2
MY6297 MATa ura3-52 leu2-3, 112 his3-⌬200 ade2-101 dsk2⌬::LEU2 rad23⌬pkc1ts
MY6296 MATa ura3-52 trp1-1 his4 ade2-101 leu2 pkc1⌬::LEU2 dsk2⌬::LEU2 rad23⌬::URA3
[pkc1tsYcp50]
MY6663 MATa ura3-52 ade2-101 leu2-3, 112 his7 trp1⌬1 cdc13-1 e
MY6739 MAT␣ura3-52 his3-⌬200 leu2-3, 112 mps2-1 [MPS2 URA3]
MY6764 MATa ura3-1/52 ade2-101/1oc can1-100? leu2-3, 112 his3-⌬200 spc110-220 [pJG109]
MY6805 MATa ura3 trp his kar1⌬17 pkc1⌬::LEU2 [pkc1tsYcp50] b
MY6809 MAT␣ura3-52 leu2 ade2-101 his3-⌬200 dsk2⌬::LEU2 rad23⌬mpk1⌬::HIS3 [MR2757]
MY6855 MATa ura3-52 ade2-101 leu2-3, 112 his3-⌬200 cdc31-1
MY6857 MATa ura3-52 ade2-101 leu2-3 112 his3-⌬200 cdc31-1 bck1⌬::HIS3
MY6859 MATa ura3-52 ade2-101 leu2-3, 112 his3-⌬200 cdc31-1 mpk1⌬::HIS3
MY6861 MAT␣ura3-52 leu2 ade2-101 his3-⌬200 dsk2⌬::LEU2 rad23⌬bck1⌬::HIS3
MY6862 MATa ura3-52 ade2-101 leu2-3, 112 trp1-⌬63 cdc31-1 slg1⌬::LEU2
MY6871 MAT␣ura3-52 leu2 lys2-801 GAL2⫹his3-⌬200 trp1⌬63 spc110-220 slg1⌬::LEU2
TABLE 2
(Continued)
Strain Genotype Source
MY6877 MATa ura3-1/52 ade2-101/1oc can1-100? leu2-3, 112 his3-⌬200 spc110-220 bck1⌬::HIS3
MY6880 MATa ura3-1/52 ade2-101/1oc can1-100? leu2-3, 112 his-3-⌬200 spc110-220 mpk1⌬::HIS3
MY6903 MAT␣ura3-52 his3-⌬200 leu2-3, 112 mpk1⌬::HIS3 mps2-1 [MPS2 URA3]
MY6974 MATa ura3-52 ade2-101 leu2-3, 112 his3-⌬200 cdc31-1swi4⌬::HIS3 [MR2012]
SLJ127 MATa ura3-1 ade2-1 can1-100 leu2-3, 112 his3-11 trp1-1 cdc15-2 f
V.370 MAT␣ura3-52 leu2-3, 112 his3-⌬200 mps2-1 g aJohn Diffley, Imperial Cancer Research Fund, United Kingdom.
bD. E. Levin, Johns Hopkins University, Baltimore. cT. Davis, University of Washington, Seattle. dBreck Byers, University of Washington, Seattle. eVirginia Zakian, Princeton University, Princeton. fD. O. Morgan, University of California, San Francisco. gMark Winey, University of Colorado, Boulder.
Spc110p fusion (Stirlinget al. 1994). Protein extracts were ture (data not shown). We also tested whether the
down-prepared by trichloroacetic acid (TCA) precipitation stream components of the PKC1 pathway, BCK1-20 and (Wright et al. 1989). Protein extracts were separated on a
MPK1 2, could suppress dsk2⌬rad23⌬, kar1-⌬17, and 6.5% SDS-PAGE as described (Laemmli1970). A 1:1000
dilu-cdc31-2. As shown in Figure 1, BCK1-20 was a suppressor tion of affinity-purified Spc110p antibodies was used for
West-ern blotting. of kar1-⌬17 but not of the other mutations. High-copy MPK1 was a very weak suppressor of kar1-⌬17. All high-copy suppression results are summarized in Figure 1D. RESULTS
These results show that overexpression of multiple com-ponents of the PKC1 pathway can suppress mutations
Overexpression of members of thePKC1/MPK1
path-way suppresses SPB duplication mutants: Mutations in in all of the genes that are known to affect the first step
in SPB duplication. Strikingly, the upstream compo-CDC31, KAR1, DSK2, and RAD23 result in defects at an
early step during SPB duplication. Specifically, cdc31, nents of the PKC1 pathway, PKC1 and SLG1, were consis-tently the strongest suppressors.
kar1-⌬17, and dsk2⌬rad23⌬mutants arrest in G2/M as
large-budded cells with a monopolar spindle and a sin- Slg1p suppresses the SPB duplication defects through
Pkc1p:High-copy SLG1 was a better suppressor of
kar1-gle SPB (Byers1981;Vallenet al. 1992;Bigginset al.
1996). All combinations of dsk2⌬, rad23⌬, and kar1⌬17 ⌬17 than high-copy PKC1. On the other hand both were equally good suppressors of cdc31-2. We therefore mutants can be suppressed by high-copy CDC31,
sug-gesting that Cdc31p is downstream in this pathway. To wanted to determine whether SLG1 suppresses the SPB defects through PKC1 or via an independent pathway. identify other genes that interact with the CDC31-related
network of SPB duplication genes, we screened for high- We tested whether PKC1 lies downstream of SLG1 for suppression of SPB duplication mutants by asking copy suppressors of the temperature sensitivity of dsk2⌬
rad23⌬ (see materials and methods). As expected, whether high-copy PKC1 can suppress a dsk2⌬rad23⌬ slg1⌬ triple mutant. The triple mutant showed a the most frequent high-copy suppressor was CDC31. In
addition, we identified SLG1/WSC1 and PKC1, as partial stronger temperature-sensitive growth defect than the dsk2⌬ rad23⌬ double mutant. Nevertheless, high-copy suppressors of the growth defect of dsk2⌬ rad23⌬ at
restrictive temperatures (Figure 1A). SLG1/WSC1 en- PKC1 suppressed both the dsk2⌬ rad23⌬ slg1⌬ triple mutant and the dsk2⌬rad23⌬double mutant well (Fig-codes a plasma membrane protein that signals to Pkc1p
via Rho1p (Vernaet al. 1997;Jacobyet al. 1998). Pkc1p ure 2A). Therefore, Pkc1p does not require Slg1p to suppress dsk2⌬ rad23⌬ consistent with PKC1 being activates the MAP kinase module consisting of Bck1p,
Mkk1p/Mkk2p, and Mpk1p (LeeandLevin1992;Irie downstream of SLG1. We next tested whether Slg1p required Pkc1p for suppression. This experiment is et al. 1993;Leeet al. 1993).
To determine whether the suppression by high-copy complicated by the fact that PKC1 is an essential gene. We therefore determined whether high-copy SLG1 SLG1 and PKC1 was relevant to SPB duplication, we
examined their ability to suppress mutations in SPB could suppress a dsk2⌬rad23⌬pkc1tstriple mutant. Once
again, the temperature sensitivity of the dsk2⌬rad23⌬ components that show genetic interactions with dsk2⌬
rad23⌬. As shown in Figure 1, B and C, high-copy SLG1 pkc1tsmutant was more severe than that of either dsk2⌬
rad23⌬or pkc1ts. We found that high-copy SLG1 did not
and PKC1 also partially suppressed kar1-⌬17 and cdc31-2.
Two other alleles of CDC31, cdc31-1 and cd31-5, were suppress the dsk2⌬ rad23⌬ pkc1ts triple mutant at 37⬚
tempera-at 32⬚, and high-copy SLG1 may be suppressing by en-hancing the partial activity. Alternatively, suppression at 32⬚may be due to a function of Slg1p that is partially independent of Pkc1p.
Overexpression of Pkc1p pathway components does
not suppress G2/M-arrested mutants in general: The
SPB duplication mutations that showed genetic interac-tions with the PKC1 pathway cause a G2/M arrest at the restrictive temperature. Therefore, we considered the possibility that the PKC1 pathway may simply be re-quired for maintaining the integrity of large-budded cells at G2/M. If so, activation of the pathway may simply suppress the growth defect of SPB mutants by sup-pressing the loss of viability of G2/M-arrested cells. To address this possibility, we examined whether high-dos-age SLG1 or PKC1 could rescue the temperature sensitiv-ity of a variety of mutants that arrest at G2/M through different mechanisms. We examined cdc13-1, defective for telomere metabolism (WoodandHartwell1982; HartwellandSmith1985;Garviket al. 1995), cdc15-2, defective in APC activation in M phase (Jaspersen et al. 1998), and cdc7-1, defective for DNA replication (NjagiandKilbey 1982;Sclafaniet al. 1988). More-over, to address whether SPB mutants may generally interact with the PKC1 pathway, we tested two other SPB duplication mutants not known to be linked to Cdc31p, spc110-220 (Sundberget al. 1996) and mps2-1 (Wineyet al. 1991). We found that cdc13-1, mps2-1, cdc15-2, or cdc7-1 strains were not suppressed by 2 SLG1 or 2 PKC1 (Figure 3). Therefore, we conclude that overexpression of PKC1 pathway genes do not generally suppress mutants that arrest in G2/M.
Another trivial possibility is that overexpression of PKC1 pathway components suppresses SPB duplication
Figure1.—Overexpression of the PKC1 pathway suppresses mutants by extending the length of G1 and thereby the temperature sensitivity of SPB duplication mutants. (A) allowing extra time for SPB duplication to occur. To
dsk2⌬ rad23⌬ (MY4193), (B) kar1-⌬17 (MS2083), and (C) test this possibility, we monitored cell cycle progression cdc31-2 (MY2260), bearing the indicated plasmids were
spot-in straspot-ins either overexpressspot-ing or deleted for PKC1 ted onto SC-ura in four 10-fold serial dilutions and incubated
pathway components after release from various synchro-at different tempersynchro-atures. Shown are plsynchro-ates synchro-at 23⬚ and 37⬚
(left to right, A) and 23⬚, 35⬚, and 37⬚ (left to right, B and nizations. We found that in all cases the cells proceeded C). (D). Summary diagram showing suppression of the SPB through the cell cycle with equivalent kinetics ( Ivanov-duplication mutants by plasmids containing PKC1 pathway
skaandRose2000, and data not shown). Therefore, the genes. The PKC1 pathway genes are listed in the order of the
PKC1 pathway does not suppress the SPB duplication genetic pathway, with SLG1 being the most upstream. Solid
mutants by altering progression through G1. lines represent partial suppression and broken lines represent
very weak suppression. We found that SLG1 and PKC1 overex- SPC110is genetically linked to theCDC31network of
pression suppressed all four SPB duplication mutants (data SPB duplication genes: Strikingly, the spc110-220 mu-for spc110-220 suppression shown in Figure 3). In addition, tant strain was strongly suppressed by high-copy PKC1
BCK1-20, and to a lesser extent overexpression of MPK1,
sup-and SLG1 (Figure 3). This observation raised the possi-pressed kar1-⌬17. BCK1-20 and MPK1 2 did not suppress
bility that Spc110p may have functions related to those cdc31-2 or dsk2⌬rad23⌬mutants. The ability of BCK1-20 and
MPK1 2plasmids to suppress spc110-220 was not tested. of Kar1p, Cdc31p, or Dsk2p and Rad23p. Spc110p is a component of the inner SPB plaque that binds calmodu-lin (Geiser et al. 1993; Kilmartin et al. 1993). The spc110-220 mutation leads to defective SPB assembly at Pkc1p’s essential function. However, we found that, at
an intermediate temperature of 32⬚, high-copy SLG1 the restrictive temperature ostensibly because of re-duced interaction between Spc110p and calmodulin did suppress the dsk2⌬ rad23⌬ pkc1ts triple mutant as
Figure 2.—(A) PKC1 overex-pression suppresses dsk2⌬rad23⌬ slg1⌬. The suppression of dsk2⌬ rad23⌬was examined in the pres-ence (top) or abspres-ence (bottom) of SLG1. dsk2⌬rad23⌬(MY4193) and dsk2⌬rad23⌬slg1⌬(MY5689) strains were transformed with the indicated plasmids and spotted as in Figure 1. The dsk2⌬ rad23⌬ slg1⌬ grew more poorly than
dsk2⌬rad23⌬at both 30⬚and 37⬚. PKC1 overexpression suppressed
the temperature sensitivity of both strains, suggesting that SLG1 was not required for suppression. (B)
SLG1 overexpression partially
sup-pressed dsk2⌬rad23⌬pkc1ts. To
de-termine whether suppression of
dsk2⌬rad23⌬by SLG1 overexpres-sion requires PKC1, we con-structed a dsk2⌬ rad23⌬ pkc1ts
(MY6297) and transformed it with the indicated plasmids. While
SLG1 overexpression did not
sup-press dsk2⌬rad23⌬pkc1tsat 37⬚, it
partially suppressed at 32⬚.
no evidence for in vivo interaction has been reported. sion of spc110-220 must occur by a mechanism different from that of kar1-⌬17. These results suggest that Cdc31p To test whether spc110-220 shares characteristics with
kar1-⌬17 and dsk2⌬rad23⌬, we asked whether spc110- also interacts with Spc110p in vivo, consistent with the in vitro data. In summary, increased dosage of Pkc1p 220 could be suppressed by high-copy CDC31 or
CDC31-16, known suppressors of dsk2⌬ rad23⌬ and kar1-⌬17 and Slg1p suppressed all four mutations that can be suppressed by high-copy Cdc31p. These findings suggest (Vallenet al. 1994; Bigginset al. 1996); see Figure 4.
We found that high-copy CDC31, but not CDC31-16, that the PKC1 pathway interacts with one or several proteins in the Cdc31p SPB assembly network.
partially rescued spc110-220 (Figure 4). In contrast,
mps2-1, an SPB mutant that is not suppressed by overex- Mechanism of high-dosage suppression: We next wanted to investigate how high-dosage SLG1 and PKC1 pression of the Pkc1p pathway, was also not suppressed
by any of the plasmids. Therefore, we concluded that suppressed the growth defect of the SPB mutants. The simplest model is that the PKC1 pathway contributes to high-copy CDC31 suppressed spc110-220 specifically.
Be-cause CDC31-16 did not suppress spc110-220, suppres- SPB function. However, the PKC1 pathway has a
well-Figure 3.—High-copy
SLG1 or PKC1 is not a
gen-eral suppressor of G2/M-arrested mutants. The
spc110-220 mutant was strongly
suppressed by overexpres-sion of SLG1 or PKC1. However, none of the other mutants were suppressed by these plasmids. Tempera-ture-sensitive mutant strains
spc110-220 (HSY11-4d),
cdc13-1 (MY6663), mps2-1
(v.370), cdc15-2 (SLJ127),
cdc7-1 (2032) were
Figure4.—High-dosage Cdc31p specifically suppresses the temperature sensitivity of
spc110-220. The spc110-220 mutant was
sup-pressed by high-copy CDC31, but not by single-copy CDC31 or CDC31-16. In contrast, kar1-⌬17 was suppressed by high-copy CDC31 and CDC31-16, while mps2-1 was not suppressed by
any plasmid. Strains kar1-⌬17 (MS2083), spc110-220 (HSY11-4d), and mps2-1 (v.370)
were transformed with vector (pRS426),
CDC31 CEN (MR2012), CDC31 2(MR2032), corresponding wild-type plasmid, or
CDC31-16 CEN (MR2223). Ura⫹transformants were spotted as in Figure 1 and incubated for 2 days at the permissive temperature (30⬚) shown at the bottom and restrictive temperatures (37⬚ for kar1-⌬17 and spc110-220 and 35⬚ for
mps2-1), shown at the top.
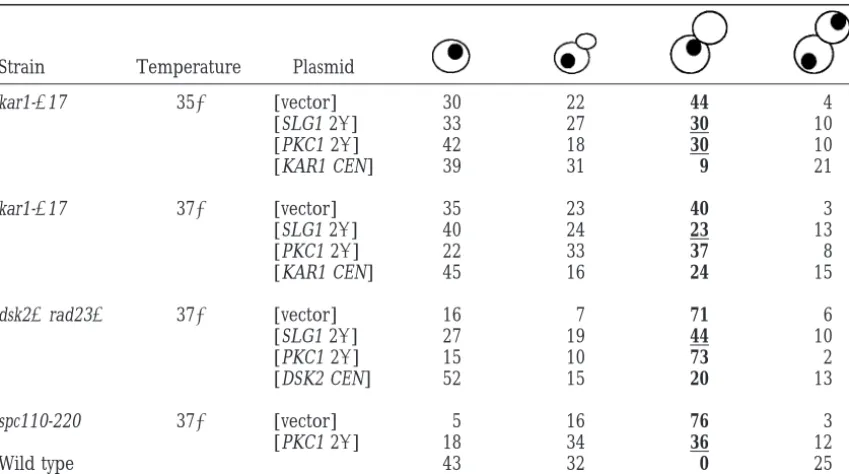
established role in cell-wall integrity. Therefore, there We next tested whether high-dosage SLG1 and PKC1 suppressed the G2/M arrest of kar1-⌬17, dsk2⌬rad23⌬, is a formal possibility that genes in the CDC31 network
play an additional role in cell-wall integrity and that and spc110-220 mutants, caused by the failure in SPB duplication. The G2/M arrest is demonstrated by large-these SPB mutants suffer cell-wall defects that can be
suppressed by SLG1 and PKC1. The compromised cell budded cells with a single nucleus, indicating failure of the duplicated DNA to segregate. If high-dosage SLG1 walls in PKC1 pathway mutants cause cell lysis and death.
We therefore tested whether the kar1-⌬17 and dsk2⌬ and PKC1 suppressed the SPB duplication defects of the mutants, we would expect a decrease in the percent-rad23⌬mutants exhibited cell lysis, using FUN-1, a
fluo-rescent viability probe (Millardet al. 1997). The major- age of G2/M-arrested cells. Indeed, SLG1 and PKC1 overexpression partially suppressed the G2/M arrest de-ity of pkc1tsmutant cells (98%) were dead after 6 hr at
the nonpermissive temperature of 37⬚ (Table 3). In fect of kar1-⌬17 at 35⬚ (from 44 to 30%) and SLG1 overexpression partially suppressed at 37⬚ (from 40 to contrast, under similar conditions, we found only a small
percentage of dead cells in the dsk2⌬rad23⌬and kar1- 23%) (Table 4), consistent with the partial suppression of the temperature sensitivity of kar1-⌬17. Similarly, the ⌬17 strains at the restrictive temperature (8 and 16%
respectively) and these were not affected by high-copy G2/M defect of dsk2⌬rad23⌬was partially suppressed by high-copy SLG1 at 37⬚ (from 71 to 44%; Table 4). SLG1 or high-copy PKC1 (Table 3). These results
indi-cate that decreased lysis/cell death is not likely to be Moreover, high-copy PKC1 suppressed the G2/M arrest phenotype of spc110-220 (from 76 to 36%). Therefore, the mechanism by which SLG1 and PKC1 suppress the
SPB duplication mutants. SLG1 and PKC1 partially suppressed the G2/M arrest of kar1-⌬17, dsk2⌬rad23⌬, and spc110-220 mutants, im-plying that they suppress the SPB duplication defects.
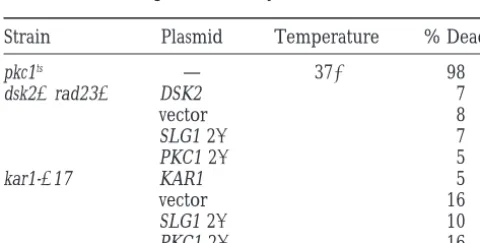
TABLE 3 Our findings support the idea that SLG1 and PKC1
provide a positive function for SPB duplication. SPB duplication mutations do not cause a
Synthetic lethal interactions between SPB duplication
significant cell lysis defect
mutants and multiple members of thePKC1 pathway:
Strain Plasmid Temperature % Dead If the Pkc1p pathway does regulate SPB duplication, then PKC1 pathway mutations should be partially
com-pkc1ts — 37⬚ 98
promised for SPB duplication. If so, their function
dsk2⌬rad23⌬ DSK2 7
should be revealed by synthetic lethal interactions with
vector 8
the relevant SPB mutants. In an independent genetic
SLG1 2 7
PKC1 2 5 screen, we identified mutations in MPK1, the PKC1
path-kar1-⌬17 KAR1 5 way MAP kinase, as being synthetically lethal with
kar1-vector 16 ⌬17 (seematerials and methods). In Figure 5A, we
SLG1 2 10
demonstrate synthetic lethality by the inability of a strain
PKC1 2 16
to segregate a URA3-based plasmid containing a wild-Mutant strains dsk2⌬ rad23⌬ (MY4193) and kar1⌬-17 (MS- type copy of one of the genes mutated on the chromo-2083) transformed with indicated plasmids and a pkc1tsstrain
some. kar1-⌬17 and mpk1⌬ all grew on 5-FOA, a wild-(DL523) were grown at 23⬚to early log phase and then
trans-type control, at 23⬚. In contrast, the mpk1⌬ kar1-⌬17 ferred to 37⬚ for 6 hrs. Death was assayed using the FUN-1
double mutant strain was extremely sensitive to 5-FOA dye (seematerials and methods). This experiment was
TABLE 4
Activation ofPKC1pathway partially suppresses the G2/M arrest of SPB duplication mutants
Strain Temperature Plasmid
kar1-⌬17 35⬚ [vector] 30 22 44 4
[SLG1 2] 33 27 30 10
[PKC1 2] 42 18 30 10
[KAR1 CEN] 39 31 9 21
kar1-⌬17 37⬚ [vector] 35 23 40 3
[SLG1 2] 40 24 23 13
[PKC1 2] 22 33 37 8
[KAR1 CEN] 45 16 24 15
dsk2⌬rad23⌬ 37⬚ [vector] 16 7 71 6
[SLG1 2] 27 19 44 10
[PKC1 2] 15 10 73 2
[DSK2 CEN] 52 15 20 13
spc110-220 37⬚ [vector] 5 16 76 3
[PKC1 2] 18 34 36 12
Wild type 43 32 0 25
SPB mutant strains were transformed with the indicated plasmids and grown at 23⬚ in SC-ura and then shifted to 35⬚or 37⬚. Cell cycle distribution was analyzed after 6 hr at 35⬚or 4 hr at 37⬚for kar1-⌬17 (MS2083),
after 8 hr at 37⬚for dsk2⌬rad23⌬(MY4193), and after 6 hr at 37⬚ for spc110-220 (HSY11-4d) and wild type (MS1554). Cell cycle distribution was analyzed as described inmaterials and methods. More than 100 cells were counted for each strain. Numbers in table represent percentages. The experiment was repeated several times with reproducible results. Boldface type indicates the aberrant G2/M class of cells. Underlined numbers indicate instances where the G2/M class is significantly reduced relative to the vector control.
for survival. Thus, the mpk1⌬kar1-⌬17 double mutant synthetically lethal, while the other combinations had restrictive temperatures lower than either parent. Figure combination is synthetically lethal.
We next asked whether the synthetic lethality ob- 5C summarizes all of the exacerbated growth defects and synthetic lethal interactions observed. On the basis served between mpk1 and kar1-⌬17 could be extended
to other SPB components and members of the PKC1 of both high-dosage suppression and synthetic lethal genetic interactions, we conclude that the PKC1 pathway pathway. First, to test whether other SPB mutants
showed synthetic lethal interactions with mpk1⌬, we de- provides a function that is crucial for survival of the SPB duplication mutants.
leted MPK1 in the dsk2⌬ rad23⌬, cdc31-1, and
spc110-220 mutant strains containing the appropriate wild-type Characterization of the double mutants: The genetic interactions presented thus far do not distinguish gene on URA3 plasmids. The mpk1⌬ was synthetically
lethal with dsk2⌬rad23⌬and spc110-220 mutations (Fig- whether the synthetic defects affect SPB duplication per se. Although this seems most likely, an alternative is that ure 5A). The cdc31-1 mpk1⌬ double mutant showed a
greatly exacerbated growth defect at temperatures per- mutations in SPB components could have secondary defects in cell integrity that are exacerbated in the ab-missive for either single mutant (Figure 5B). In contrast,
the mps2-1 mpk1⌬ double mutant did not show a syn- sence of the PKC1 pathway. To assess the nature of the synthetic defects directly, we analyzed the phenotypes thetic lethal phenotype (Figure 5A) confirming that the
synthetic lethal interactions are restricted to the CDC31 of the double mutant strains. This analysis is technically compromised by the fact that the terminal phenotype network of SPB duplication genes. In summary, all the
CDC31-related SPB mutants analyzed showed either syn- associated with a defect in SPB duplication (arrest at G2/M with a large bud and an unduplicated SPB) re-thetic lethality or synre-thetic growth defects when MPK1
was deleted. quires an extended period of bud growth. Bud growth
is extremely sensitive to defects in the PKC1 pathway Next, we tested whether mutations in other
compo-nents of the PKC1 pathway showed synthetic lethal inter- causing cells to lyse before the large bud stage. Thus, at the restrictive temperature, most double mutants ar-actions with the SPB duplication mutants. All the double
mutant combinations tested showed synthetic lethality rested as a mixture of small-budded and large-budded cells and so were not informative as to whether there or severely exacerbated growth defects (Figure 5, A and
Figure5.—Synthetic lethality and synthetic growth defects between SPB mutants and mutations in the PKC1 pathway. (A)
mpk1⌬is synthetically lethal with kar1-⌬17, dsk2⌬rad23⌬, and spc110-220, but not with mps2-1. In addition, bck1⌬is synthetically lethal with kar1-⌬17. kar1-⌬17 mpk1⌬: (I) kar1-⌬17 mpk1⌬(MS5886), (II) mpk1⌬(MS5930), (III) kar1-⌬17 (MS2373), and (IV)
wild type (MS1554). kar1-⌬17 bck1⌬: (I) kar1-⌬17 bck1⌬(MS5888), (II) bck1⌬(MS5938), (III) kar1-⌬17 (MS5820), and (IV) wild
type (MS1554). dsk2⌬rad23⌬mpk1⌬: (I) dsk2⌬rad23⌬mpk1⌬(MY6809), (II) mpk1⌬(MS6547), (III) dsk2⌬rad23⌬(MY5064), and (IV) wild type (MS1554). spc110-220 mpk1⌬: (I) spc110-220 mpk1⌬(MY6880), (II) mpk1⌬(MS6547), (III) spc110-220 (MY6764), and (IV) wild type (MS6556). mps2-1 mpk1⌬: (I) mps2-1 mpk1⌬(MY6903), (II) mpk1⌬(MS6547), (III) mps2-1 (MY6739), and (IV) wild type (MS1554). Strains growing on SC-ura contain a URA3 plasmid bearing the appropriate SPB duplication gene. (B) Exacerbation of growth defects of SPB mutants in the absence of PKC1 pathway members. In each case, we observed that the double or triple mutants had lower restrictive temperatures than their parents. Strains were spotted on synthetic complete or YPD at permissive temperature of 23⬚or 30⬚(left) and restrictive temperature (right). Shown from top to bottom: slg1⌬(MY4455),
kar1-⌬17 (MS2083), slg1⌬kar1-⌬17 (MY4646), dsk2⌬rad23⌬(MY4193), slg1⌬dsk2⌬rad23⌬(MY4916), cdc31-1 (MY2259), slg1⌬ cdc31-1 (MY6862), pkc1ts(DL523), kar1-⌬17 (MS2083), pkc1tskar1-⌬17 (MY6805), dsk2⌬rad23⌬ (MY4193), pkc1tsdsk2⌬rad23⌬
(MY6296), bck1⌬(MS 5938), dsk2⌬rad23⌬(MY4193), bck1⌬dsk2⌬rad23⌬(MY6861), cdc31-1 (MY6855), bck1⌬cdc31-1 (MY6857), mpk1⌬(MS6547), cdc31-1 (MY6855), and mpk1⌬cdc31-1 (MY6859). (C) Summary diagram showing synthetic lethal interactions
(solid line) or synthetic growth defects (broken line) between PKC1 pathway and SPB duplication mutants. PKC1 pathway mutations are listed in order of the genetic pathway, with slg1⌬being the most upstream mutation in the pathway. Note that the growth phenotype of pkc1tscdc31-1 and pkc1tsspc110-220 double mutants were not tested.
shown). Synchronizing the cultures in G1 with␣-factor, growing mpk1⌬ kar1-⌬17 mutant propagated at 23⬚ showed an increase in G2/M-arrested cells (53%) rela-followed by release from arrest, did not alleviate the
problem (data not shown). However, mpk1⌬exhibited a tive to the kar1-⌬17 single mutant (33%) without show-ing an elevated frequency of dead cells (Table 5). Thus, strong synthetic lethality with kar1-⌬17 at a temperature
TABLE 5
Thekar1-⌬17 mpk1⌬shows an increase in large-budded, G2/M-arrested cells
Strain Temperature % dead
Wild type 23⬚ 60 36 0 4 1
kar1-⌬17 25 42 33 0 3
mpk1⌬ 63 34 1 2 16
kar1-⌬17 mpk1⌬ 33 14 53 0 5
Analysis of the cell cycle distribution was performed as described inmaterials and methods. More than 100 cells were counted for each strain. The numbers represent percentages of total number of cells counted. The experiment was repeated several times with reproducible results. Boldface indicates significant enhancement of the aberrant G2/M class relative to the single mutants.
consistent with the hypothesis that the PKC1 pathway tions that compromise the ability of CDC31-16 and DSK2-1 to suppress kar1-⌬17 (W. Khalfanand M. Rose, plays a positive role in SPB duplication.
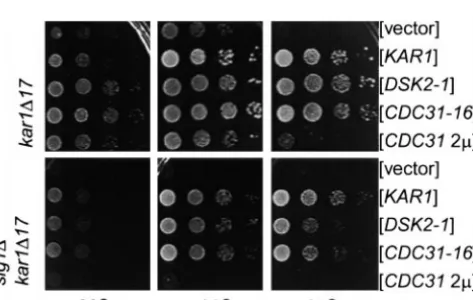
Pkc1p pathway activity is required for proper Cdc31p unpublished observations; Biggins et al. 1996). As
shown in Figure 6, CDC31-16 or DSK2-1 plasmids did
functionin vivo:The genetic interactions suggest that
Pkc1p pathway activity is required for optimal function not suppress the slg1⌬kar1-⌬17 as well as KAR1 at 37⬚. Moreover, high-copy CDC31 completely failed to sup-of SPB duplication genes. All sup-of the genes suppressed
by PKC1 overexpression can also be suppressed by high- press the double deletion even at 30⬚(Figure 6, bottom). These results can be interpreted in two ways. First, Slg1p copy CDC31. Therefore, we next asked whether the
PKC1 pathway is required for Cdc31p to suppress the may be required for optimal function of Cdc31p. By this scheme the PKC1 pathway would lie upstream or SPB mutations. Specifically, we tested whether
high-copy CDC31 and the dominant alleles CDC31-16 and in a parallel pathway leading to activation of Cdc31p. Alternatively, Cdc31p itself may be required to activate DSK2-1 could suppress kar1-⌬17 in the absence of the
Pkc1p pathway component Slg1p. The dominant alleles the PKC1 pathway. In this scenario, the PKC1 pathway would lie downstream of Cdc31p, and mutations in cdc31 CDC31-16 and DSK2-1, but not high-copy CDC31, can
suppress a complete deletion of KAR1 (Vallen et al. and the other SPB duplication genes would be predicted to alter PKC1 pathway activity. We tested this second 1994) and suppress kar1-⌬17 as well as KAR1 (Figure 6,
top). Previous analysis has failed to uncover any muta- idea by measuring PKC1 pathway activity in the SPB duplication mutants in a number of ways. First, as a measure of MKK1 and MKK2 activity, we tested whether MPK1 was phosphorylated in a kar1-⌬17 strain upon mild heat shock treatment (Kamadaet al. 1995;Verna et al. 1997). We found that Mpk1p’s ability to be phos-phorylated was not compromised in the kar1-⌬17 strain (data not shown). We also examined the in vitro kinase activity of Pkc1p immunoprecipitated from a kar1-⌬17 strain, using myelin basic protein (MBP) as the substrate (Watanabe et al. 1994). Again, we found no evidence that Pkc1p kinase activity was affected, at least in vitro (data not shown). Finally, using a LacZ-reporter assay system, we examined the transcriptional activity of Rlm1p, a MADS-box transcription factor whose activity depends on Mpk1p phosphorylation (Watanabeet al.
Figure6.—The ability of high-copy CDC31 to suppress kar1- 1997). Although Rlm1p transcriptional activity was in-⌬17 is compromised in the absence of SLG1. Tenfold serial
deed lower in the kar1-⌬17 strain, this was a general dilutions of MS2373, MY3752-3755 (top) and MY4642
trans-effect of G2/M arrest, since another G2/M mutant, formed with indicated plasmids (bottom) were plated on
SC-cdc13, also showed reduced levels of Rlm1p transcrip-ura plates and grown for 2 days at the indicated temperatures.
The top shows that kar1-⌬17 is temperature sensitive and that tional activity. Taken together, it seems unlikely that CDC31-16 and DSK2-1 strongly suppressed the temperature the CDC31 pathway activates the PKC1 pathway. We
sensitivity whereas CDC31 overexpression was a partial
suppres-therefore interpret the inability of high-copy CDC31 to sor. The bottom shows that in the absence of SLG1, the
sup-suppress the kar1-⌬17 slg1⌬double mutant (Figure 6) pressing ability of DSK2-1 and CDC31-16 are partially
spindles (Friedman et al. 1996; Stirling and Stark 1996). The SPB duplication gene Mps1p is one of the kinases responsible for Spc110p phosphorylation (M. Wineyand T. Davis, unpublished communication). On the basis of the time of the appearance of the p120 form and analysis of cdc mutants, the phosphorylation that produces the p120 form does not appear to be
Figure7.—Spc110p phosphorylation is defective in pkc1ts
required for SPB duplication (Friedman et al. 1996). and mpk1⌬mutants. Protein extracts were prepared from asyn- We reasoned that the phosphorylation state of Spc110p chronously growing,␣-factor-arrested, or nocodazole-arrested
could serve as a useful marker for earlier event(s) in wild-type strain (MS1554) at 30⬚and asynchronously growing
SPB duplication. We therefore examined the phosphor-pkc1ts(DL523) and MPK1⌬strains (MS5930) at 30⬚or 37⬚(see
materials and methods). For ␣-factor arrest, MS1554 was ylation of Spc110p in asynchronous wild-type, pkc1ts, and
treated with 10g/ml␣-factor for 3 hr at 30⬚. For nocodazole mpk1⌬cultures grown in YPD at 30⬚and shifted to 37⬚ arrest, MS1554 was treated with 5g/ml nocodazole for 3 hr for 3 hr. An asynchronous wild-type culture grown at 37⬚ at 30⬚. The top arrow marks the p120 form of Spc110p and
contained both p120 and p112 forms,␣-factor-arrested the bottom arrow marks the p112 form. The p120 form is
wild-type strain contained the faster migrating p112 absent in the pkc1tsmutant at restrictive temperature (37⬚)
and is also greatly diminished in the mpk1⌬ mutant at 30⬚ form, and nocodazole-arrested wild-type cells contained
and 37⬚. the p120 form (Figure 7 and Friedman et al. 1996;
StirlingandStark1996). In contrast to the wild-type strain, the p120 form was absent in the pkc1tsmutant at
the slg1⌬mutant. The observation that CDC31-16 and
37⬚. Similarly, an mpk1⌬culture showed reduced levels DSK2-1, but not high-copy CDC31, could partially
sup-of p120 at permissive temperature (30⬚) and drastically press the kar1-⌬17 slg1⌬double mutant at 37⬚suggests
reduced levels of p120 at 37⬚. that the dominant suppressors may act by a different
The disappearance of the p120 form in pkc1ts and
mechanism that is partially independent of the PKC1
mpk1⌬strains is not likely to be due to the accumulation pathway. In support of the idea that they act differently,
of cells at a stage in the cell cycle prior to Spc110p CDC31 2, but not CDC31-16, suppressed the
tempera-phosphorylation. First, although the pkc1tsmutant
accu-ture-sensitive growth defect of spc110-220 (Figure 4).
mulates small-budded cells, the small-budded cells have
Phosphorylation of Spc110p is defective inpkc1tsand
already initiated DNA replication and formed short
mpk1⌬mutants: We next sought to explore the
molecu-spindles (LevinandBartlett-Heubusch1992; Ivan-lar mechanism by which the PKC1 pathway affects SPB
ovskaandRose2000). Therefore, on the basis of the duplication. One possibility is that the PKC1 pathway
pkc1tsmutant phenotype, we would not expect a block
phosphorylates an SPB component. On the basis of the
in the production of the p120 form due to cell cycle genetic interactions, possible candidates include Kar1p,
arrest. Second, in the case of the mpk1⌬ mutant, the Cdc31p, and Spc110p. We first examined Kar1p in the
strain did not arrest at any particular stage of the cell different PKC1 pathway mutants and found no change
cycle at the restrictive temperatures. We directly exam-in the mobility of various Kar1p forms (data not shown).
ined the cell cycle distribution of pkc1tsand mpk1⌬strains
We next tested Spc110p, which exists as at least two
from aliquots taken at the time of protein extract prepa-distinct phosphorylated forms of molecular weights 112
ration. As shown in Table 6, while there were differences kD and 120 kD. The 120-kD form (p120) arises from
in their cell cycle distribution compared to wild type, additional serine/threonine phosphorylation of the
the distribution was not correlated with the presence 112-kD form and is cell cycle regulated, predominating
in small-budded cells with duplicated DNA and short or absence of the p120 form. These results suggest that
TABLE 6
Cell cycle distribution ofpkc1tsandmpk1⌬strains
Strain Temperature
Wild type 37⬚ 43 37 20 0
pkc1ts 30⬚ 51 39 8 2
37⬚ 23 47 29 1
mpk1⌬ 30⬚ 31 35 34 0
37⬚ 32 27 41 0
Wild type (MS1554), pkc1ts(DL523), and mpk1⌬(MS5930) strains were grown at 23⬚in YPD and then shifted
tion factor complex consisting of Swi4p/Swi6p (SBF) has also been suggested to be a second downstream effector of MPK1 (Maddenet al. 1997). Swi4p and Swi6p physically associate with Mpk1p, and phosphorylation of Swi4p and Swi6p is dependent on Mpk1p (Maddenet al. 1997). These results suggest that Swi4/Swi6 complex may be a target of PKC1 pathway activity. However, SBF activity is also thought to be required to turn on the PKC1 pathway via Cdc28p/Clns (Mazzoni et al. 1993; Mariniet al. 1996;Zarzovet al. 1996;Grayet al. 1997). To determine whether Rlm1p and/or Swi4p are impor-tant for the survival of the SPB muimpor-tants, we generated kar1-⌬17 rlm1⌬and kar1-⌬17 swi4⌬double mutants con-taining KAR1 URA3 plasmids and examined their ability to lose the plasmid on 5-FOA at a range of temperatures. As shown in Figure 8A, we found that the kar1-⌬17 swi4⌬ strain was synthetically lethal, similar to the kar1-⌬17 mpk1⌬double mutant. In contrast, the kar1-⌬17 rlm1⌬ double mutant did not grow any more poorly than ei-ther single mutant alone at a range of temperatures (Figure 8B). Since the cdc31-1 mpk1⌬ double mutant also has a severe growth defect, we examined whether cdc31-1 mutants were sensitive to mutations in either SWI4 or RLM1. Similar to the results obtained with the kar1-⌬17 swi4⌬double mutant, a cdc31-1 swi4⌬double mutant was synthetically lethal, whereas a cdc31-1 rlm1⌬ double mutant did not show any synthetic growth
de-Figure8.—Synthetic lethality of kar1-⌬17 and cdc31-1 with fects (Figure 8, C and D). These results argue that swi4⌬, but not rlm1⌬. The top represents SC-ura plates while Swi4p’s function is crucial for survival of the SPB
dupli-the bottom represents 5-FOA plates. (A) (I) kar1-⌬17 swi4⌬
cation mutants, kar1-⌬17 and cdc31-1, in activating the (MS6944), (II) swi4⌬(MS6934), (III) kar1-⌬17 (MS5820), and
PKC1 pathway and/or transmitting a signal downstream (IV) wild type (MS1554) all contain KAR1 URA3 (MR76 or
of Mpk1p. Clearly however, Rlm1p is not the target of MR77). (B) (I) kar1-⌬17 rlm1⌬(MS6947), (II) rlm⌬(MS6937),
(III) kar1-⌬17 (MS5820), and (IV) wild type (MS1554) all Mpk1p activity that is relevant for SPB duplication. contain KAR1 URA3 (MR76 or MR77). (C) (I) cdc31-1 swi4⌬ Because Swi4p might mediate its effects through tran-(MY6974), (II) swi4⌬(MS6934), (III) cdc31-1 (MY3883), and
scriptional regulation, and because Cdc31p function is (IV) wild type (MS1554) all contain CDC31 URA3 (MR2012).
compromised in the kar1-⌬17 slg1⌬mutant (Figure 6), (D) (I) cdc31-1 rlm1⌬ (II) rlm1⌬ (MS6937), (III) cdc31-1
one possible explanation for the genetic interactions (MY3883), and (IV) wild type (MS1554) all contain CDC31
URA3 (MR2012). would be if the PKC1 pathway regulated CDC31
tran-scription. To address this question, we examined the levels of CDC31 mRNA in asynchronously growing wild-the lack of phosphorylation is not due to a block in cell type, pkc1ts, mpk1⌬, and swi4⌬mutants either at 23⬚or
cycle progression. Taken together, our results suggest 37⬚. We found that the levels of CDC31 mRNA were that Pkc1p and/or Mpk1p may directly or indirectly similar in the mutant and wild-type strains (data not phosphorylate Spc110p. These results may provide a shown). We also found the levels of Cdc31p protein in mechanistic basis for the genetic interactions between pkc1tsmutant to be comparable to that of wild type at
thePKC1 pathway and SPB duplication genes. 37⬚. In addition, the levels of Kar1p and Spc110p were
Swi4p but not Rlm1p function is required in SPB wild type in pkc1tsand mpk1⌬strains (data not shown).
duplication mutants:Having established that Pkc1p and Therefore, it is unlikely that the PKC1 pathway regulates
the MAP kinase module affect SPB duplication, we the synthesis of Cdc31p, Kar1p, or Spc110p. It remains wanted to identify the downstream effectors of the PKC1 possible, however, that the expression of an unidentified pathway that transduce the signal to the SPB. One well- SPB component is regulated by the PKC1 pathway. established downstream target of Mpk1p is Rlm1p, a
MADS-box family transcription factor whose activity is
DISCUSSION MPK1 dependent (Watanabeet al. 1995, 1997;Dodou
andTreisman1997). However, unlike mpk1⌬, deletion Genetic interactions with SPB duplication genes
sug-of RLM1 does not result in cell morphogenesis and/or gest a role for the PKC1 pathway in SPB duplication:
transcrip-PKC1 signal transduction cascade. Overexpression of duplication mutants. Recently, a genome-wide analysis has identified numerous genes that are transcriptionally SLG1 or PKC1 partially rescued the temperature
sensitiv-ity of dsk2⌬rad23⌬, kar1-⌬17, and cdc31-2. We assume upregulated or downregulated in response to MPK1 activity (JungandLevin1999). The transcriptional reg-that overexpression leads to activation of the PKC1
path-way. The suppression was specific to SPB duplication ulation of most of the genes identified in the genome analysis was mediated by Rlm1p, and all genes code defects and was not due to general suppression of G2/M
arrest. High-dosage SLG1 and PKC1 did not simply res- for components of the cell-wall biosynthesis machinery (JungandLevin1999). Since loss of Mpk1p function cue cell-wall-related defects of the SPB mutants, because
we did not find significant cell lysis in dsk2⌬ rad23⌬ results in a much more severe growth defect than the loss of Rlm1p, Mpk1p must have other targets as well. and kar1-⌬17. It is interesting that the most upstream
members of the PKC1 pathway, SLG1 and PKC1, were It is possible that other genes that are transcriptionally regulated by Mpk1p were missed in the genome analysis. the strongest suppressors. We present two
interpreta-tions of this observation. First, the PKC1 pathway may However, the results of Jung and Levin (1999) raise the interesting possibility that Mpk1p activates its targets branch at one or more points and the different
compo-nents of the PKC1 pathway may suppress the SPB dupli- post-transcriptionally. Indeed, the transcript levels of CDC31 and protein levels of Cdc31p, Kar1p, and cation mutants by different mechanisms. Alternatively,
consistent with the observations of other researchers, Spc110p are normal in the PKC1 pathway mutants, sug-gesting that the pathway may regulate SPB compo-overexpression of upstream components may activate
the PKC1 pathway more strongly than overexpression nent(s) post-translationally.
Formally, the PKC1 pathway may function upstream, of downstream components, resulting in different levels
of suppression of the SPB duplication mutants (Gray in parallel, or downstream of the CDC31 pathway. If the PKC1 pathway is downstream, then mutations in the et al. 1997).
We also observed somewhat different behaviors SPB duplication genes may affect the level of PKC1 pathway activity. We tested this hypothesis using several among the different PKC1 pathway components in our
synthetic lethal analyses. The severity of the interactions different markers for PKC1 pathway activity and in all cases we found that mutations in SPB duplication genes was the reverse of the suppression, with the most
down-stream components showing the most severe synthetic did not alter the activity of the PKC1 pathway. Therefore, it is unlikely that the CDC31 pathway is upstream of the phenotypes in combination with SPB duplication
muta-tions. The SPB mutants were particularly sensitive to PKC1 pathway.
Less clear is whether the PKC1 functions upstream mutations in MPK1. With the exception of cdc31, all
the SPB mutants showed synthetic lethality with mpk1⌬, or in parallel with the CDC31 pathway. We found that Slg1p is required for Cdc31p’s SPB function (Figure while mutations in the other PKC1 pathway genes
re-sulted in viable strains with more severe growth defects. 6). This result suggests that the PKC1 pathway affects Cdc31p, either by activating Cdc31p directly or by acti-In addition, the mpk1⌬ kar1-⌬17 double mutant had
an exacerbated G2/M arrest phenotype, suggesting a vating other components with which Cdc31p interacts at the SPB. Given that Spc110p phosphorylation was requirement for the MPK1 gene in SPB duplication. It
is possible that Mpk1p is crucial for the survival of SPB defective in pkc1tsand mpk1⌬mutants and that Spc110p
functionally interacts with Cdc31p, Spc110p may well mutants because it is targeted and activated by other
MAP kinase signaling pathways, especially in the ab- be one of the targets of PKC1 pathway activity. However, given the timing of the appearance of p120, it is unlikely sence of upstream components of the PKC1 pathway.
The presence of such cross-talk may help to explain why that this particular phosphorylation is the relevant regu-latory event. The p112 form has not been resolved from mutations in upstream components do not exacerbate
the growth defect of SPB duplication mutants as severely the unphosphorylated form of the protein (Friedman et al. 1996). Therefore, it is possible that the p120 ap-as mutations in MPK1.
The SPB mutants cdc31 and kar1 showed synthetic pears as a consequence of earlier phosphorylation(s) of Spc110p. In such a scenario, the lack of p120 could lethality with swi4⌬ arguing that, like Mpk1p, Swi4p is
also crucial for the survival of the SPB duplication mu- be an indication that earlier phosphorylation events relevant to SPB duplication have not occurred in the tants. The Swi4p/Swi6p transcription factor complex
(SBF) is a proposed target of Mpk1p (Madden et al. pkc1tsand mpk1⌬mutants.
While our data suggest that the PKC1 pathway is in-1997). However, SBF is also an activator of the PKC1
pathway via the Cdc28p/Clns (Grayet al. 1997). There- volved in SPB duplication, pkc1 mutants have not been reported to exhibit defects in SPB duplication. The fore, while the synthetic lethal results argue that Swi4p
is as crucial as Mpk1p, they do not discriminate whether slg1⌬ strain does have a phenotype that can be taken as including an SPB defect; in the cold, the mutant fails the upstream or downstream activity of Swi4p is
impor-tant. The lack of genetic interactions between the SPB to initiate SPB duplication even though it buds and eventually lyses (IvanovskaandRose2000). The possi-mutants and rlm1⌬ clearly rule out the possibility that