1071-412X/03/$08.00⫹0 DOI: 10.1128/CDLI.10.6.982–989.2003
Copyright © 2003, American Society for Microbiology. All Rights Reserved.
Modern Complement Analysis
Michael Kirschfink
1* and Tom E. Mollnes
2Institute of Immunology, University of Heidelberg, Heidelberg, Germany,1and Institute of Immunology, The National Hospital,
University of Oslo, Oslo, Norway2
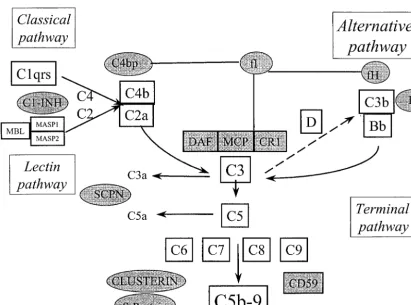
Complement, as a vital part of the body’s immune system, provides a highly effective means for the destruction of invad-ing microorganisms and for immune complex elimination (for reviews, see references 77 and 78). Through activation and interaction with its respective receptors on various immune cells, complement is closely linked to the adaptive immune response. A normally functioning complement system is also required for physiological tissue regeneration and repair (37). Thus, complement defects (Table 1) increase the susceptibility to infection and, if classical pathway components are affected, are frequently associated with autoimmune disorders (11). Protection against complement-mediated tissue destruction is provided by various soluble and membrane-bound regulatory proteins (Fig. 1) (51). Defects in this regulation may therefore lead to serious disorders due to unrestricted activation. In addition, as a key mediator of inflammation, complement also significantly contributes to tissue damage in various clinical disorders (50, 76).
The present paper briefly reviews clinical indications for complement analysis and gives an overview on current meth-ods in routine and experimental complement diagnostics.
THE COMPLEMENT SYSTEM
Complement is activated by three pathways (Fig. 1), the classical pathway, the alternative pathway, and the recently discovered lectin pathway (14), all of which lead to the forma-tion of the cytolytic membrane attack complex, C5b-9. Follow-ing complement activation, the biologically active peptides C5a and C3a elicit a number of proinflammatory effects, such as chemotaxis of leukocytes, degranulation of phagocytic cells, mast cells, and basophils, smooth muscle contraction, and in-crease of vascular permeability (29). Upon activation by these complement products, the inflammatory response is further amplified by subsequent generation of toxic oxygen radicals, induction of synthesis, and release of arachidonic acid metab-olites and cytokines. Consequently, an (over)activated comple-ment system presents a considerable risk of harming the host by directly and indirectly mediating inflammatory tissue de-struction.
Under physiological conditions, activation of complement is effectively controlled by the coordinated action of soluble and membrane-associated regulatory proteins (51). Soluble com-plement regulators, such as C1 inhibitor, C4b-binding protein (C4bp), factors H and I, clusterin, and S-protein (vitronectin), restrict the action of complement in body fluids at multiple
sites of the cascade reaction (Fig. 1). In addition, each indi-vidual cell is protected against the attack of homologous com-plement by surface proteins, such as the comcom-plement receptor 1 (CD35) and the membrane cofactor protein (CD46), as well as by the glycosylphosphatidylinositol-anchored proteins, de-cay-accelerating factor (DAF; CD55), and CD59. There is increasing evidence that these protection mechanisms are aug-mented on malignant cells, leading to their refractoriness against complement (26).
CLINICAL INDICATIONS FOR COMPLEMENT ANALYSIS
Clinical and experimental evidence underlines the promi-nent role of complement in immunodeficiency disorders (Ta-ble 1) and in the pathogenesis of numerous inflammatory dis-eases (11, 50, 76), including immune complex and autoimmune disorders such as systemic lupus erythematosus (SLE) (79) and autoimmune arthritis (34). The following are disorders associ-ated with complement activation: systemic inflammatory reac-tion syndrome, multiple organ dysfuncreac-tion syndrome, isch-emia-reperfusion syndrome, angioedema, capillary leak syndrome, hyperacute and acute graft rejection, vasculitis, ne-phritis, autoimmune disorders (e.g., SLE, rheumatoid arthritis, and myasthenia gravis), biomaterial incompatibility (e.g., fol-lowing dialysis or cardiopulmonary bypass), and severe trauma, burn, and sepsis. Only recently has complement also been implicated in neurodegenerative disorders, such as Alzhei-mer’s disease (52), multiple sclerosis (70), and Guillain-Barre´ syndrome (62). Complement activation after polytrauma sub-stantially contributes to the development of systemic inflam-matory reaction syndrome and multiple organ failure (82). In recent years, complement has been recognized as a major effector mechanism of reperfusion injury (35, 46, 48). Further-more, activation of complement is a critical event in the patho-genesis of sepsis and septic shock (10, 15).
The inflammatory response induced by artificial surfaces in hemodialysis and extracorporeal circuits may lead to organ dysfunction. Here, complement activation has been shown to be associated with transient neutropenia, pulmonary vascular leukostasis, and occasionally, anaphylactic shock of variable severity in patients undergoing hemodialysis or cardiopulmo-nary bypass (27, 47).
In recent years, great progress has been made in comple-ment analysis to better define disease severity, evolution, and response to therapy. Modern diagnostic technologies which focus on the quantification of complement-derived split prod-ucts or protein-protein complexes now provide a comprehen-sive insight into the activation state of the system.
(i) Recurrent infections.Complement defects (Table 1) may
be the cause of immunodeficiency-mediated recurrent infec-* Corresponding author. Mailing address: Institute of Immunology,
University of Heidelberg, Im Neuenheimer Feld 305, 69120 Heidel-berg, Germany. Phone: 49 06221 56 4076. Fax: 49 06221 56 5586. E-mail: [email protected].
982
on August 17, 2020 by guest
http://cvi.asm.org/
TABLE 1. Genetic complement de ficiencies and disease association a Component Disease condition(s) Analytical method(s) b Genetic basis(es) C1q, C1s Collagen vascular disease, SLE, bacterial infections CH50, RID, ELISA, C1q/C1s function, SDS-PAGE Various point mutations; acquired form due to C1q autoantibodies MBL Bacterial infections MBL function, ELISA Various point mutations C2 Neisserial infections, respiratory infections, SLE (often symptomless) CH50, RID, ELISA, C2 function, SDS-PAGE Type I, 28-bp deletion at exon-intron junction 6 leading to splicing error; linked to HLA-A25-B18-C2Q0-C4A4,B2-DR2; type II, impaired C2 secretion C3 Bacterial infections, glomerulonephritis CH50, RID, ELISA, C3 function, electrophoresis Various independent mutations in the C3 gene; 800-bp deletion due to Alu repeat-induced mutation; acquired due to C3 nephritic factor C4A/B Collagen vascular disease, autoimmune diseases (e.g., SLE, autoimmune hepatitis, scleroderma) CH50, RID, ELISA, SDS-PAGE, electrophoresis with hemolytic overlay 1. Complete gene deletion; 2. gene conversion (isotype change); 3. nonexpression due to 2-bp insertion 3 stop codon C5 Recurrent neisserial infections CH50, RID, ELISA, C5 function Point mutation 3 stop codon C6 Recurrent neisserial infections CH50, RID, ELISA, C6 function Partial de ficiency: splice site mutation 3 loss of exons 16 and 17 (3 ⬘ truncation) C7 Recurrent neisserial infections CH50, RID, ELISA, C7 function Various independent point mutations and 1-bp deletions 3 stop codons, substitutions, splice site mutations, deletion of exons 7 and 8 C8 (C8 ␣ – ␥ and C8  ) Recurrent neisserial infections, C8 function CH50, ELISA, SDS-PAGE C8B gene: point mutation in exon 9 3 stop codon (common), also other rare mutations; C8A gene: splice site mutation in intron 6 3 10-bp insertion and premature stop codon C9 Recurrent neisserial infections, SLE (often symptomless) RID, ELISA, C9 function Point mutations 3 stop codon; cysteine substitution P Fulminant neisserial infections, sepsis AH50, ELISA Various point mutations 3 stop codons, frameshifts, amino acid substitution Factor B Only heterozygotes detected, clinically inapparent AH50, RID, ELISA, SDS-PAGE Unknown Factor H Neisserial infections, HUS RID, ELISA (reduced C3 levels), SDS-PAGE 4-bp deletion 3 stop codon, point mutations 3 stop codon, cysteine substitutions (impaired secretion), various point mutations Factor I Meningitis, pyogenic infections CH50, RID, ELISA (reduced C3 levels and no C3d detectable) Evidence for Alu repeat-induced mutation C1 inhibitor HAE C1 inhibitor function, RID, ELISA (reduced C1, C4, and C2 levels) Various point mutations and small deletions, exon deletions due to Alu repeat recombinations; acquired autoimmune form DAF, CD59 Paroxysmal nocturnal hemoglobinuria Acidic lysis test, FACS analysis GPI anchor defect due to PIG-A gene point and frameshift mutations c CR3 (CD11b/CD18) Recurrent bacterial skin infections FACS analysis De ficiency of common  chain a Table adapted from reference 68 with permission of the publisher. b CH50, complement hamolytic activity; AH 50 ,alternative pathway hamolytic activity; RID, radial immunodif fusion; SDS-PAGE, sodium dodecyl sulfate-polyacrylamide gel electrophoresis; F ACS, fluorescence-activated cell sorter. c GPI, glycosylphosphatidylinositol; PIG-A, phosphatidylinositol glycan-A (gene).
on August 17, 2020 by guest
http://cvi.asm.org/
tions (11). Recurrent neisserial infections, particularly if sys-temic and with rare serotypes, are associated with complement defects, preferentially in the terminal C5-C9 pathway. The frequency of meningococcal disease reported in C5-C9-defi-cient individuals reflects a 1,000- to 10,000-fold-higher risk in this population (65). Late-component-deficient patients, which are readily identified by assays of total complement function should be vaccinated with tetravalent vaccines against all four strains ofNeisseria meningitidis(A,C,W135, and Y) (49)
There is also a high prevalence of properdin deficiency in patients with meningococcal disease, mainly caused by uncom-mon serogroups W135 and Y, with a fatality rate of more than 25% (33). The most common type I deficiency is characterized by the absence of properdin in plasma, whereas in type II deficiency, properdin is low but detectable (⬍10% of the nor-mal level). However, properdin defects are not always detected by the conventional hemolytic assay (AH50). Thus, direct quantification of properdin or the application of a lipopolysac-charide (LPS)-based alternative pathway activation enzyme-linked immunosorbent assay (ELISA) (see below) may be in-dicated in such cases.
Mannose binding lectin (MBL) is a key protein of the lectin activation pathway of complement. Genetic deficiency of MBL is rather frequent and is associated with increased infection risk (28), particularly in the mother-child window age of 6 to 18 months. MBL should therefore be added to the list of param-eters to be tested if immunodeficiency is suspected. However,
the interpretation of the results should be done with some caution since, due to genetic variants (18), concentrations of MBL per se do not fully reflect the activity of the lectin path-way. Here, recently developed functional assays (30, 59) should be included.
(ii) Autoimmune diseases.Complement defects, particularly
of the classical pathway, are frequently associated with SLE-like autoimmune disease (79). The strength of the association of a complement deficiency with systemic lupus erythematodes increases from C2 (10% prevalence) to C1r/s (57% preva-lence), C4 (75% prevalence) and C1q (90% prevalence) (61). In general, autoimmunity is only occasionally associated with a genetic complement defect. Therefore, in active SLE, par-ticularly with renal involvement, low CH50 and C4 titers are more often due to increased in vivo activation, which can be verified by the detection of complement activation products. In patients with severe clinical outcome, such as lupus nephritis, autoantibodies to C1q are often found and may be of prog-nostic value (7).
(iii) Membranoproliferative glomerulonephritis (MPGN)
and hemolytic uremic syndrome (HUS).In certain vasculitides
and kidney diseases, a substantial activation and consumption of C3 due to defective alternative pathway regulation can be observed.
Patients suffering from MPGN, especially of the histologi-cally defined type II, often show low levels of CH50, AH50, and C3. This results from a continuous C3 activation due to an FIG. 1. Schematic diagram of the complement cascade reaction. Complement regulatory proteins are shown as shaded circles (fluid phase regulators) and boxes (membrane-associated regulators). MASP, MBL-associated serine protease; C1 INH, C1 inhibitor; SCPN, serum car-boxypeptidase N; DAF, CD55; MCP, membrane cofactor protein (CD46); CR1, complement receptor 1 (CD35).
on August 17, 2020 by guest
http://cvi.asm.org/
autoantibody, termed C3 nephritic factor (C3NeF), which sta-bilizes the labile C3bBb complex (8, 72). Here, identification of C3NeF is of significant importance.
HUS is characterized by microangiopathic hemolytic ane-mia, thrombocytopenia, and acute renal failure. There is com-pelling evidence that the atypical form of this fatal disease is often associated with a mutation in the C terminus of factor H, expressed as suboptimal regulator activity and consequently leading to hypocomplementemia (83). Occasionally, factor H is also reduced in cases of MPGN type II.
(iv) HAE. Hereditary angioedema (HAE) is an autosomal
dominant condition with reduced concentration (type 1) or function (type 2) of C1 inhibitor (6, 9, 56). With respect to life-threatening consequences of edema formation, early diag-nosis of the C1 inhibitor deficiency in these patients is ex-tremely important. As spontaneous mutations can occur, a negative family history does not exclude the diagnosis. Fur-thermore, the penetrance differs significantly and minor symp-toms or subclinical cases occur. The pathophysiology of HAE is complex, but it is now generally accepted that formation of bradykinin through activation of the kallikrein-kinin system, which is also controlled by C1 inhibitor, is the major inductor of the edema (58). The diagnosis is based on C1 inhibitor and C4 quantification. It is important to include both antigenic and functional assays for C1 inhibitor, since 15% of the patients have type 2 HAE with normal or even increased antigen con-centration of C1 inhibitor. C4 is usually low in both cases, even between the attacks, and serves as a valuable supplement to C1 inhibitor analysis. The concentration of C3 is usually normal in HAE. All HAE patients described so far are heterozygous in their deficiency, with less than 50% of the normal concentra-tion and/or funcconcentra-tion. A homozygous C1 inhibitor deficiency is probably not compatible with life. Acute treatment requires purified C1 inhibitor or plasma transfusion, whereas androgens can be given prophylactically.
A clinical picture identical to HAE may occur in patients who develop autoantibodies to C1 inhibitor, in some cases related to hematologic malignancies (1). In addition to re-duced C1 inhibitor and C4 levels, these patients frequently have a low C1q concentration in contrast to the HAE patients. Identification of this form of acquired angioedema is impor-tant, as its acute treatment requires higher doses of C1 inhib-itor.
(v) Paroxysmal nocturnal hemoglobinuria.A somatic
mu-tation in the gene coding for the phosphatidylinositol anchor results in a decreased expression of membrane proteins linked to this structure (5), including DAF (CD55) and CD59. De-creased expression of DAF and/or CD59 renders the red cells susceptible to complement-mediated lysis, which is the hall-mark of this condition. Diagnosis is traditionally made by Ham’s test (acid lysis test) but is now specifically assessed by flow cytometric analysis of the respective cell surface proteins (31).
COMPLEMENT TESTS
(i) Functional assays. (a) Total complement activity of the
classical, lectin, and alternative pathways. Hemolytic assays
have traditionally been used to assess the functional activity of the complement system. They provide insight into the integrity
of the entire cascade reaction. These tests are particularly useful in the investigation of suspected complement deficien-cies.
Although described in numerous modifications, hemolytic assays are still based on protocols first described by Mayer (39) and Rapp and Borsos (63). Serial dilutions of the sample to be analyzed are incubated with antibody-sensitized sheep eryth-rocytes at a defined temperature. Hemolytic assays are per-formed either in tubes or in agarose plates. The results are usually expressed as reciprocal dilutions of the sample required to produce 50 or 100% lysis (CH50 or CH100, respectively). Tests evaluating the functional activity of the alternative path-way (AH50) use guinea pig, rabbit, or chicken erythrocytes as target cells (25). Here, activation of the classical pathway has to be blocked by adding EGTA to chelate Ca2⫹, and an
opti-mal concentration of Mg2⫹is required. Detection of low or
absent hemolytic activity in CH50 and/or AH50 directs further complement analysis.
However, AH50 is frequently normal or only slightly re-duced in properdin deficiencies and needs to be replaced by alternative assays such as LPS activation assay or specific pro-perdin tests (see below).
Simplified assays with a single serum dilution and a large number of erythrocytes are suitable for large-scale clinical screening, especially if they can be performed as semiauto-mated microassays (54). Commercially available cytolytic as-says replacing erythrocytes with sensitized liposomes (36) offer certain advantages with regard to logistics, reproducibility, and suitability for automated systems.
The function of complement can also be tested by measuring the deposition of activation products upon activation of the serum with immobilized complement-activating substances (immunoglobulin M [classical pathway], LPS [alternative path-way], or mannose [lectin pathway]) on a microtiter plate. The combination of detection of C9 in the immunoglobulin M ELISA, properdin deposition in the LPS ELISA, and C4 bind-ing in the lectin pathway assay excludes a suspected comple-ment deficiency (12, 23, 59). It is likely that these tests will replace hemolytic assays in the future.
(b) Activity of individual components.Titration of individual
complement proteins requires the addition of those compo-nents which are needed to complete the reaction sequence. A correct estimation of the components’ activity is obtained by calculation of functional molecules (63). The most convenient way to detect the functional activity of individual complement components is to test for the sample’s capability to reconstitute the hemolytic activity of a serum which is deficient for the protein. With the availability of complement-deficient or -de-pleted human sera, titration of individual complement proteins became much easier to perform. To finally prove that only one single component is lacking, a purified functionally active com-ponent can be added to the serum to restore hemolytic activity of the respective pathway.
Hemolytic assays are sensitive to in vitro activation of com-plement in serum. If serum is heat inactivated, has been stored for a long time at room temperature, or contains complement-activating agents (e.g., immune complexes or cold agglutinins), the hemolytic activity is reduced and may even be zero. Thus, fresh serum (⬍48 h old) should be used or samples should be stored at⫺70°C until tested.
on August 17, 2020 by guest
http://cvi.asm.org/
(c) C1 inhibitor activity. Fifteen percent of patients with HAE have a dysfunctional protein (type 2 HAE) with normal or increased concentrations of C1 inhibitor antigen. To diag-nose this form of HAE, it is mandatory to include a functional C1 inhibitor test (53, 81), which can be done by commercially available quantitative chromogenic assays.
(ii) Immunochemical assays for individual components.
In-dividual complement components, irrespective of functional activity, can be measured by immunoprecipitation tests (radial immunodiffusion [RID] or nephelometer techniques), ELISA, or Western blotting. In routine diagnosis, C3 and C4 (and sometimes factor B) are most frequently measured, followed by C1 inhibitor, to verify the diagnosis of type 1 HAE. MBL and properdin quantification should now be considered in-cluded in the investigation of a suspected immunodeficiency. In most cases, where total hemolytic activities (CH50 and AH50) indicate a complement deficiency, immunochemical as-says can be performed as an alternative to functional asas-says for the individual components. If a defect is verified immuno-chemically, further functional assays are not required. On the other hand, if immunochemical assays do not reveal any defi-ciency, the component may be functionally inactive and only a functional assay can verify the diagnosis.
(iii) Analysis of complement activation products.Total
he-molytic activity and levels of individual complement compo-nents may also give some indication of an ongoing complement activation, e.g., low levels C4 and CH50 in active SLE. How-ever, the same results may be obtained in cases with a genetic deficiency of components, e.g., C4, as frequently observed in SLE. Furthermore, in vivo complement activation in conjunc-tion with an acute-phase reacconjunc-tion may leave the individual components within the normal range despite ongoing con-sumption, since most of the complement components are acute-phase proteins. In general, total hemolytic activity and individual component measurements are useful as first-level screening techniques, but they are not sensitive enough to detect pathologically increased complement activation in vivo. During the last two decades, highly specific monoclonal anti-bodies have been produced which recognize neoepitopes only exposed upon activation-induced conformational changes (42). This enables direct capture of the activation fragment by ELISA (44) or to high-capacity immunosorbents (16) without interference of the nonactivated component. These assays have today replaced the older generation of tests, which were ham-pered by preassay precipitation or fractionation steps or are time-consuming or of low capacity.
Complement activation products may be either split frag-ments after enzymatic cleavage of certain components, e.g., C4 (C4a and C4d), C3 (C3a and C3d), factor B (Bb), and C5 (C5a), or protein complexes, like C1rs-C1 inhibitor, the prop-erdin-containing alternative pathway convertase C3bBbP, and SC5b-9 (soluble C5b-9 bound to S protein), where activated components are bound to their respective regulators.
Neoepitope-specific assays to detect fluid-phase activation have been described and are partly commercially available for the classical pathway (C1rs-C1 inhibitor, C4d, and C4bc), the alternative pathway (Ba, Bb, and C3bBbP), C3 (C3a, iC3b/ C3bc, and C3d) and the terminal reaction sequence (C5a and SC5b-9). Many of the neoepitope-specific antibodies can also
be used to detect in situ complement activation by applying immunohistochemistry.
Complement activation products are usually present in only trace amounts in vivo, but they are rapidly generated in vitro (41). Therefore, it is crucial that samples are collected and stored properly to avoid in vitro activation. Blood should be drawn directly into EDTA-containing tubes at a final EDTA concentration of at least 10 mM. Alternatively, nafamostat mesilate can be used as an anticoagulant (60), whereas citrate and heparin do not sufficiently block complement activation. The sample should be cooled, and plasma should be prepared immediately and stored at⫺70°C.
The various complement activation products have different half-lives in vivo. This is important for the activation product(s) of choice to be measured. Due to rapid receptor binding, the biologically highly active and important C5a fragment has a half-life of approximately 1 min (57) and is difficult to detect in samples obtained in vivo, whereas the various C3 activation products are readily detectable due to half-lives of a few hours (74). The half-life of SC5b-9 is 50 to 60 min (45). SC5b-9, in contrast to C3 activation products, is relatively stable in vitro and is a reliable indicator of terminal pathway activation.
The amount of an activation product should be related to the concentration of the native component, since a low level of native component would yield smaller amounts of activation products during in vivo activation. Thus, it has been postulated that the ratio between the activation product and the native component is a more sensitive indicator of in vivo activation than the activation product alone (55).
(v) Assays for complement-binding autoantibodies.C3NeF
can be measured in a decay experiment by stabilization of the alternative pathway convertase C3bBb (8, 24). A semiquanti-tative screening assay based on a stable cell (sheep erythro-cyte)-bound convertase was reported by Rother (67). Fluid-phase conversion of C3 upon the mixture of normal serum and C3NeF-containing patients’ serum can be visualized in an im-munofixation assay (also commercially available).
Autoantibodies to C1q or to C1 inhibitor can be detected by ELISA with purified proteins immobilized on the microtiter plate. Interpretation of results from various dilutions of pa-tients’ serum or plasma should be done in comparison with data received from a large panel of healthy controls (71). In a C1q autoantibody ELISA, sample dilution requires the pres-ence of a high salt concentration to avoid immune complex binding.
COMPLEMENT ANALYSIS IN EXPERIMENTAL SETTINGS
(i) In vitro experiments with human serum and blood.
Ac-tivation mechanisms and intervention of complement activa-tion can readily be studied in vitro with human serum. Since there are no cells present and there is no biological turnover, any activation product including C5a can be detected. A more physiological approach, however, is to use whole human blood. In this case, anticoagulants need to be added. However, most of the anticoagulants, such as EDTA, citrate, and heparin interfere with complement activation and should be avoided. From our own experience, recombinant hirudin (lepirudin), a highly specific thrombin inhibitor, does not affect complement
on August 17, 2020 by guest
http://cvi.asm.org/
and seems particularly useful for complement studies with whole human blood (40).
To avoid further activation during preparation and storage after the experiments have been performed, it is necessary to add EDTA (optimal in combination with nafamostat mesilate or futhan) at the end of the experiments (60).
In vivo, experimental studies with human complement are limited for obvious reasons. However, certain clinical condi-tions can be mimicked by experimental settings, e.g., extracor-poreal circulation (hemodialysis or cardiopulmonary bypass), and have proved useful for the study of complement activation and possible effects of complement intervention.
(ii) Animal experiments. (a) Application of gene-deficient or
knockout animals.A number of animals with genetic
deficien-cies of individual complement components have been de-scribed (66) and are commercially available (e.g., C2-, C4-, or C3-deficient guinea pigs, C3-deficient dogs, C5-deficient mice, and C6-deficient rats and rabbits). During the last few years, an additional number of knockout mice have been generated (e.g., deficient for C1q, C3, C4, and factor B) (2). These ani-mals are particularly useful for the study of the importance of complement to various diseases and for delineating activation mechanisms and the pathways of activation involved. In prin-cipal, reconstitution experiments with purified components should be included, but for practical reasons, this is not always possible, since limited numbers of components have been pu-rified from the different species. The addition of a component from another species may be valuable, with a functional cross-reactivity provided. Administration of a purified human com-ponent to a gene-deficient mouse may work with respect to some complement effects, such as lysis, but molecular incom-patibility may imply that different receptor-mediated effects cannot be studied. Reconstitution with whole serum of the same species may serve as an alternative.
(b) Complement depletion.Cobra venom factor (CVF) has
been used for decades to decomplement animals. CVF is anal-ogous to C3b and activates C3 but is resistant to regulation by human factors I and H (75). CVF treatment is frequently and misleadingly called inhibition of complement; in fact, an ex-treme activation with subsequent complement consumption takes place. The animals usually tolerate this activation (fairly) well, since only a limited activation of the terminal pathway occurs. Nevertheless, adverse effects from CVF have been re-ported (38, 64), and one should be very cautious with the interpretation of the data from such experiments. A direct inhibition of complement at a certain level is recommended instead of CVF treatment when mechanisms of complement activation are to be studied.
(c) Testing animal complement. Hemolytic assays can be
designed for any species. However, information obtained from such assays are limited but still valuable. They can be used to monitor the effect of complement inhibition by a certain sub-stance or as a semiquantitative measure of complement acti-vation under controlled conditions, since hemolysis will be lowered proportionally to the consumption of complement. In many animal species, a comprehensive analysis of complement is still hampered by the lack of suitable reagents and assays. A major factor affecting the evaluation of complement activity in the serum of previously unstudied species is the choice of an appropriate target cell. Low or nondetectable classical pathway
activity of murine or bovine complement, for example, results from applying conventional methods with sensitized sheep erythrocytes. Comparison of classical pathway activity between different species is difficult to evaluate, since hemolytic titers are also influenced by the source and quality of erythrocyte-specific antibodies (19). Applying optimal test conditions, it can be demonstrated that virtually all species of laboratory and domestic animals possess classical as well as alternative path-way activity (3, 21, 73). The compatibility of components within the cascade sequence between different species and guinea pigs or humans often allows the titration of individual compo-nents (3, 80). To evaluate the pathophysiological contribution of complement to certain diseases, assessment of complement activation by immunochemical means is indispensable. How-ever, as measurement of components is not a sensitive and reliable indicator of a slight activation, activation products should be tested. Whereas a number of neoepitope specific assays have been described for human complement activation, only a few are available for animal studies. These are either assays for human activation products which cross-react with other species or assays particularly designed for animal studies. The latter include assays based on monoclonal antibodies to neoepitopes of guinea pig C5a (32) and C3 activation products (17), rabbit C5a (4), and rat C5b-9 (69). Several of the human neoepitope-specific assays cross-react sufficiently well with other species, like baboon C3bc (20); baboon C3bc, C5a, and C5b-9 (43); baboon C4d, C3a, Bb, and C5b-9 (13); and porcine C5b-9 (22).
The possible species cross-reactivity of a certain assay made for human complement activation can easily be checked by activating the serum from the respective species by, e.g., zy-mosan, and comparing it to the nonactivated control. If the signal in activated serum is stronger than in the nonactivated serum, the assay cross-reacts with this species and the degree of cross-reactivity can be estimated.
REFERENCES
1. Alsenz, J., K. Bork, and M. Loos.1987. Autoantibody-mediated acquired deficiency of C1 inhibitor. N. Engl. J. Med.316:1360–1366.
2. Barrington, R., M. Zhang, M. Fischer, and M. C. Carroll.2001. The role of complement in inflammation and adaptive immunity. Immunol. Rev.180:5– 15.
3. Barta, O.1978. Testing of hemolytic complement components in domestic animals. Am. J. Vet. Res.39:1303–1308.
4. Bergh, K., and O. J. Iversen.1989. Measurement of complement activation in rabbit plasma or serum using monoclonal antibodies against C5a. Scand. J. Immunol.29:333–341.
5. Boccuni, P., L. Del Vecchio, R. Di Norto, and B. Rotoli.2000. Glycosyl phosphoinositol (GPI)-anchored molecules and the pathogenesis of parox-ysmal nocturnal hemoglobinuria. Crit. Rev. Oncol. Hematol.33:25–43. 6. Cicardi, M., L. Bergamaschini, M. Cugno, A. Beretta, L. C. Zingale, M.
Colombo, and A. Agostoni.1998. Pathogenetic and clinical aspects of C1 inhibitor deficiency. Immunobiology199:366–376.
7. Coremans, I. E., P. E. Spron, H. Bootsma, M. R. Daha, E. A. van der Voort, L. Kater, F. C. Breedveld, and C. G. Kallenberg.1995. Changes in antibodies to C1q predict renal relapses in systemic lupus erythematosus. Am. J. Kidney Dis.26:595–601.
8. Daha, M. R., D. T. Fearon, and K. F. Austen.1976. C3 nephritic factor (C3NeF): stabilization of fluid phase and cell-bound alternative pathway convertase. J. Immunol.116:1–7.
9. Davis, A. E.1988. C1 inhibitor and hereditary angioneurotic edema. Annu. Rev. Immunol.6:595–628.
10. de Boer, J. P., A. A. Creasey, A. Chang, D. Roem, A. J. Eerenberg, C. E. Hack, and F. B. Taylor, Jr.1993. Activation of the complement system in baboons challenged with liveEscherichia coli: correlation with mortality and evidence for a biphasic activation pattern. Infect. Immun.61:4293–4301.
11. Figueroa, J. E., and P. Densen.1991. Infectious diseases associated with complement deficiencies. Clin. Microbiol. Rev.4:359–395.
12. Frederikson, G. N., L. Truedsson, and A. G. Sjoeholm.1993. New procedure
on August 17, 2020 by guest
http://cvi.asm.org/
for the detection of complement deficiency by ELISA. Analysis of activation pathways and circumvention of rheumatoid factor influence. J. Immunol. Methods166:263–270.
13. Fung, M., P. G. Loubser, A. Undar, M. Mueller, C. Sun, W. N. Sun, W. K. Vaughn, and C. D. Fraser, Jr.2001. Inhibition of complement, neutrophil, and platelet activation by an anti-factor D monoclonal antibody in simulated cardiopulmonary bypass circuits. J. Thorac. Cardiovasc. Surg.122:113–122. 14. Gadjeva, M., S. Thiel, and J. C. Jensenius.2001. The mannan-binding-lectin
pathway of the innate immune system. Curr. Opin. Immunol.13:74–78. 15. Hack, C. E., J. H. Nuijens, R. J. Felt-Bersma, W. O. Schreuder, A. J.
Eerenberg-Belmer, J. Paardekooper, W. Bronsveld, and L. G. Thijs.1989. Elevated plasma levels of the anaphylatoxins C3a and C4a are associated with a fatal outcome in sepsis. Am. J. Med.86:20–26.
16. Hartmann, H., B. Lubbers, M. Casaretto, W. Bautsch, A. Klos, and J. Kohl.
1993. Rapid quantification of C3A and C5A using a combination of chro-matographic and immunoassay procedures. J. Immunol. Methods166:35–44. 17. Hawlisch, H., M. A. Z. Vilsendorf, W. Bautsch, A. Klos, and J. Kohl.2000. Guinea pig C3 specific rabbit single chain Fv antibodies from bone marrow, spleen and blood derived phage libraries. J. Immunol. Methods236:117–131. 18. Hibberd, M. L., M. Sumiya, J. A. Summerfield, R. Booy, and M. Levin.1999. Association of variants of the gene for mannose-binding lectin with suscep-tibility to meningococcal disease. Meningococcal Research Group. Lancet
353:1049–1053.
19. Higgins, D. A., and D. J. Langley.1985. A comparative study of complement activation. Vet. Immunol. Immunopathol.9:37–51.
20. Hiramatsu, Y, N. Gikakis, J. H. Gorman III, M. M. Khan, C. E. Hack, H. A. T. Velthuis, L. Sun, C. Marcinkiewicz, A. K. Rao, S. Niewiarowski, R. W. Colman, L. H. Edmunds, Jr., and H. L. Anderson III.1997. A baboon model for hematologic studies of cardiopulmonary bypass. J. Lab. Clin. Med.
130:412–420.
21. Ish, O., G. L. Ong, N. Desai, and M. J. Matthes.1993. The specificity of alternative complement pathway-mediated lysis of erythrocytes: a survey of complement and target cells from 25 species. Scand. J. Immuol.38:113–122. 22. Jansen, J. H., K. Hogasen, and T. E. Mollnes.1993. Extensive complement activation in hereditary porcine membranoproliferative glomerulonephritis type-II (porcine dense deposit disease). Am. J. Pathol.143:1356–1365. 23. Jaskowski, T. D., T. B. Martins, C. M. Litwin, and H. R. Hill.1999.
Com-parison of three different methods for measuring classical pathway comple-ment activity. Clin. Diagn. Lab. Immunol.6:137–139.
24. Jelezarova, E., M. Schlumberger, S. Sadallah, P. J. Spa¨th, J. A. Schifferli, and H. U. Lutz.2001. A C3 convertase assay for nephritic factor functional activity. J. Immunol. Methods251:45–52.
25. Joiner, K. A., A. Hawinger, and J. A. Gelfand.1983. A study of optimal reaction conditions for an assay of the human alternative complement path-way. Am. J. Clin. Pathol.79:65–72.
26. Jurianz, K., S. Ziegler, H. Garcia-Schu¨ler, S. Kraus, O. Bohana-Kashtan, Z. Fishelson, and M. Kirschfink.2000. Complement resistance of tumor cells: basal and induced mechanisms. Mol. Immunol.36:929–939.
27. Kirschfink, M., B. Kovacs, and K. Mottaghy.1993. Extracorporeal circula-tion: in vivo and in vitro analysis of complement activation by heparin-bonded surfaces. Circ. Shock40:221–226.
28. Koch, A., M. Melbye, P. Sorensen, P. Homoe, H. O. Madsen, K. Molbak, C. H. Hansen, L. H. Andersen, G. W. Hahn, and P. Garred.2001. Acute respiratory tract infections and mannose-binding lectin insufficiency during early childhood. JAMA285:1316–1321.
29. Ko¨hl, J.2001. Anaphylatoxins and infectious and non-infectious inflamma-tory diseases. Mol. Immunol.38:75–87.
30. Kuipers, S., P. C. Aerts, A. G. Sjo¨holm, T. Harmsen, and H. van Dijk.2002. A hemolytic assay for the estimation of functional mannose-binding lectin levels in human serum. J. Immunol. Methods268:149–157.
31. Kwong, Y. L., C. P. Lee, T. K. Chan, and L. C. Chan.1994. Flow cytometric measurement of glycosylphosphatidyl-inositol-linked surface proteins on blood cells of patients with paroxysmal nocturnal hemoglobinuria. Am. J. Clin. Pathol.102:30–35.
32. Link, C., H. Hawlisch, A. M. Z. Vilsendorf, S. Gyleruz, E. Nagel, and J. Kohl.
1999. Selection of phage-displayed anti-guinea pig C5 or C5a antibodies and their application in xenotransplantation. Mol. Immunol.36:1235–1247. 33. Linton, S. M., and B. P. Morgan.1999. Properdin deficiency and
meningo-coccal disease—identifying those most at risk. Clin. Exp. Immunol.118:189– 191.
34. Linton, S. M., and B. P. Morgan.1999. Complement activation and inhibi-tion in experimental models of arthritis. Mol. Immunol.36:905–914. 35. Lucchesi, B. R., and K. S. Kilgore.1997. Complement inhibitors in
myocar-dial ischemia/reperfusion injury. Immunopharmacology38:27–42. 36. Masaki, T., N. Osaka, R. Yasuda, and H. Okada.1989. Assay of complement
activity in human serum using large unilamellar liposomes. J. Immunol. Methods.123:19–24.
37. Mastellos, D., and J. Lambris.2002. Complement: more than a ’guard’ against invading pathogens? Trends Immunol.23:485.
38. Mathieson, P. W., F. J. Qasim, S. Thiru, R. G. Oldroyd, and D. B. G. Oliveira.1994. Effects of decomplementation with cobra venom factor on experimental vasculitis. Clin. Exp. Immunol.97:474–477.
39. Mayer, M. M.1961. Complement and complement fixation, p. 133–240.InE. Kabat and M. M. Mayer (ed.), Experimental immunochemistry. C. C. Thomas, Springfield, Ill.
40. Mollnes, T. E., O. L. Brekke, M. Fung, H. Fure, D. Christiansen, B. Bergs-eth, V. Videm, K. T. Lappega˚rd, J. Ko¨hl, and J. Lambris.2002. Essential role of the C5a receptor in E. coli-induced oxidative burst and phagocytosis revealed by a novel lepirudin-based human whole blood model of inflam-mation. Blood100:1869–1877.
41. Mollnes, T. E., P. Garred, and G. Bergseth.1988. Effect of time, temperature and anticoagulants on in vitro complement activation: consequences for collection and preservation of samples to be examined for complement activation. Clin. Exp. Immunol.73:484–488.
42. Mollnes, T. E., and M. Harboe.1993. Neoepitope expression during com-plement activation—a model for detecting antigenic changes in proteins and activation of cascades. Immunologist1:43–49.
43. Mollnes, T. E., H. Redl, K. Hogasen, A. Bengtsson, P. Garred, L. Speilberg, T. Lea, M. Oppermann, O. Gotze, and G. Schlag.1993. Complement acti-vation in septic baboons detected by neoepitope-specific assays for C3b/iC3b/ C3c, C5a and the terminal C5b-9 complement complex (TCC). Clin. Exp. Immunol.91:295–300.
44. Mollnes, T. E.1997. Analysis ofin vivocomplement activation, p. 78.1–78.8. InL. A. Herzenberg, D. M. Weir, L. A. Herzenberg, and C. Blackwell (ed.), Weir’s handbook of experimental immunology, 5th ed. Blackwell Science, Boston, Mass.
45. Mollnes, T. E.1985. Early- and late-phase activation of complement evalu-ated by plasma levels of C3d, g and the terminal complement complex. Complement2:156–164.
46. Mollnes, T. E., and E. Fosse.1994. The complement system in trauma-related and ischemic tissue damage: a brief review. Shock2:301–310. 47. Mollnes, T. E.1997. Biocompatibility: complement as mediator of tissue
damage and as indicator of incompatibility. Exp. Clin. Immunogenet.14:24– 29.
48. Moore, F. D., and W. T. Caine.2000. Emerging immunologic basis of reper-fusion injury. Transplant. Rev.14:158–167.
49. Morgan, B. P., and A. Orren.1998. Vaccination against meningococcus in complement-deficient individuals. Clin. Exp. Immunol.114:327–329. 50. Morgan, B. P.1990. Complement. Clinical aspects and relevance to disease.
Harcourt Brace Jovanovich, London, United Kindom.
51. Morgan, B. P., and C. L. Harris.1999. Complement regulatory proteins. Academic Press, San Diego, Calif.
52. Mukherjee, P., and G. M. Pasinetti.2000. The role of complement anaphy-latoxin C5a in neurodegeneration: implications in Alzheimer’s disease. J. Neuroimmunol.105:124–130.
53. Munkvad, S., J. Jespersen, J. Gram, K. Overgaard, and M. Ranby.1990. Effects of methylamine and heparin on a rapid chromogenic assay of C1-esterase inhibitor in plasma. Clin. Chem.36:737–741.
54. Nilsson, U. R., and B. Nilsson.1984. Simplified assays of hemolytic activity of the classical and the alternative pathways. J. Immunol. Methods72:49–59. 55. Nuernberger, W., and S. Bhakdi.1984. Plasma C3d/C3 quotient as a
param-eter for in vivo complement activation. J. Immunol. Methods74:87–91. 56. Nzeako, U. C., E. Frigas, and W. J. Tremaine.2001. Hereditary
angioede-ma—a broad review for clinicians. Arch. Intern. Med.161:2417–2429. 57. Oppermann, M., and O. Gotze.1994. Plasma clearance of the human C5a
anaphylatoxin by binding to leukocyte C5a receptors. Immunology82:516– 521.
58. Pappalardo, E., L. C. Zingale, A. Terlizzi, A. Zanichelli, A. Folcioni, and M. Cicardi.2002. Mechanisms of C1-inhibitor deficiency. Immunobiology205:
542–551.
59. Petersen, S. V., S. Thiel, L. Jensen, R. Steffensen, and J. C. Jensenius.2001. An assay for the mannose-binding lectin pathway of complement activation. J. Immunol. Methods257:107–116.
60. Pfeifer, P. H., M. S. Kawaharay, and T. E. Hugli.1999. Possible mechanisms for in vitro complement activation in blood and plasma samples: futhan/ EDTA controls in vitro complement activation. Clin. Chem.45:1190–1199. 61. Pickering, M. C., M. Botto, P. R. Taylor, P. J. Lachmann, and M. J. Walport.
2000. Systemic lupus erythematosus, complement deficiency and apoptosis. Adv. Immunol.76:227–324.
62. Putzu, G. A., D. Figarella-Branger, C. Bouvier-Labit, A. Liprandi, N. Bianco, and J. F. Pellissier.2000. Immunohistochemical localization of cytokines, C5b-9 and ICAM-1 in peripheral nerve of Guillain-Barre syndrome. J. Neu-rol. Sci.174:16–21.
63. Rapp, H. J., and T. Borsos.1970. Molecular basis of complement action. Appleton Century Crofts, New York, N.Y.
64. Regal, J. F., D. G. Fraser, D. E. Anderson, and L. E. Solem.1993. Enhance-ment of antigen-induced bronchoconstriction after intravascular comple-ment activation with cobra venom factor. Reversal by granulocyte depletion. J. Immunol.150:3496–3505.
65. Ross, S. C., and P. Denson.1987. Complement deficiency states and infec-tion: epidemiology, pathogenesis and consequences of Neisseria and other infections in an immune deficiency. Medicine63:234–273.
66. Rother, K., and U. Rother. 1986. Hereditary and acquired complement deficiencies in animals and man. Prog. Allergy39:1–7.
on August 17, 2020 by guest
http://cvi.asm.org/
67. Rother, U.1982. A new screening test for C3 nephritis factor based on a stable bound convertase on sheep erythrocytes. J. Immunol. Methods51:
101–107.
68. Schneider, P. M., and C. Rittner.1997. Complement genetics, p. 165–198.In A. Dodds and B. Sim (ed.), Complement—a practical approach. Oxford University Press, Oxford, United Kingdom.
69. Schulze, M., P. J. Baker, D. T. Perkinson, R. J. Johnson, R. F. Ochi, R. A. Stahl, and W. G. Couser.1989. Increased urinary excretion of C5b-9 distin-guishes passive Heymann nephritis in the rat. Kidney Int.35:60–68. 70. Sellebjerg, F., I. Jaliashvili, M. Christiansen, and P. Garred.1998.
Intra-thecal activation of the complement system and disability in multiple scle-rosis. J. Neurol. Sci.157:68–74.
71. Siegert, C. H., M. R. Daha, E. A. van der Voort, and F. C. Breedveld.1990. IgG and IgA antibodies to the collagen-like region of C1q in rheumatoid vasculitis. Arthritis Rheum.33:1646–1654.
72. Spitzer, R. E., E. H. Vallota, J. Foristal, E. Sudora, A. Stitzel, N. C. Davis, and C. D. West.1969. Serum C3 lytic system in patients with glomerulone-phritis. Science164:436–437.
73. Tanaka, S., S. Kitamura, and T. Suzuji.1987. Studies on the hemolytic activity of the classical and the alternative pathway of complement in various animal species. Complement4:33–41.
74. Teisner, B., I. Brandslund, N. Grunnet, L. K. Hansen, J. Thellesen, and S. E. Svehag.1983. Acute complement activation during an anaphylactoid reac-tion to blood transfusion and the disappearance rate of C3c and C3d from the circulation. J. Clin. Lab. Immunol.12:63–67.
75. Vogel, C. W., and H. J. Mu¨ller-Eberhard.1982. The cobra venom factor-dependent C3 convertase of human complement. J. Biol. Chem.257:8292– 8299.
76. Volanakis, J. E., and M. M. Frank.1998. The human complement system in health and disease. Marcel Dekker, Inc., New York, N.Y.
77. Walport, M. J.2001. Complement. Second of two parts. N. Engl. J. Med.
344:1140–1144.
78. Walport, M. J.2001. Complement. First of two parts. N. Engl. J. Med.
344:1058–1066.
79. Walport, M. J., and P. J. Lachmann.1990. Complement deficiencies and abnormalities of the complement system in systemic lupus erythematosus and related disorders. Curr. Opin. Rheumatol.2:661–663.
80. Whaley, K.1985. Measurement of complement, p. 77–139.InK. Whaley (ed.), Methods in complement for clinical immunologists. Churchill Living-stone, Edinburgh, United Kingdom.
81. Ziccardi, R. J., and N. R. Cooper.1980. Development of an immunochemical test to assess C1 inactivator function in human serum and its use for the diagnosis of hereditary angioedema. Clin. Immunol. Immunopathol.15:465– 471.
82. Zilow, G., J. A. Sturm, U. Rother, and M. Kirschfink.1990. Complement activation and the prognostic value of C3a in patients at risk of adult respi-ratory distress syndrome. Clin. Exp. Immunol.79:151–157.
83. Zipfel, P. F.2001. Hemolytic uremic syndrome: how do factor H mutants mediate endothelial damage? Trends Immunol.22:345–348.