www.orientjchem.org
An International Open Free Access, Peer Reviewed Research Journal
2017, Vol. 33, No. (1): Pg. 157-164
Physical Chemistry Properties of Fe
3O
4@
Cyclodextrin@ (12, 12) SWCNTs as a Catalyst
ZEYNAb AbbASI, MAJId MONAJJEMI*, KARIM ZARE and MASOUMEH SAYAdIAN
Department of Chemistry, Science and Research Branch, Islamic Azad University, Tehran, Iran.
*Corresponding author E-mail: [email protected] http://dx.doi.org/10.13005/ojc/330117
(Received: January 07, 2017; Accepted: February 08, 2017) AbSTRACT
Fe3O4 is used in the water gas shift reaction as a catalyst in the “Haber process”. In this
work, the physical and chemical properties of Fe3O4 @ α-Cyclodextrin @ (12, 12) SWCNTs has been investigated. Our calculations have been done in point of chemical phenomenon and electronic properties. The Magnetic behavior, Electron densities and electrical properties such as NMR Shielding, potential energies densities, energy density, ellipticity for electron densities, ELF, LOL, index of eta and finally ECP for Fe3O4 @ -Cyclodextrin@ (12, 12) SWCNTs have been calculated
and simulated in our system. Our Calculation indicate that the Fe3O4 @ -Cyclodextrin@ (12, 12) SWCNTs are suitable surfaces for Fe3O4 such silica surfaces.
Keywords:Fe3O4, Nano-Particles, electron density, (12, 12) SWCNTs, Cyclodextrin.
INTROdUCTION
The great temperature shifts catalyst (HTS) of iron oxide1-3 stabilized by chromium oxide1-5. This
chrome-iron alloys as a catalyst is diminished at chemical-reactor-start up for generating Fe3O4 from á-Fe2O34 and Cr
2O35. Fe3O4 is one of the most
important electrical conductors within conductivities considerably higher comparison to Fe2O33,4 and this
is imputed for exchange electron between two parts of the Fe(II) and Fe(III) centers1-3.
Fe3O4 family are ferromagnetic with remarkable curie temperature (858 K) and this ferromagnetism properties of Fe3O4 appears because the spin electron of the Fe(II) and Fe(III) ions are coupled together in the octahedral structures and the spin of the Fe(III) ion are coupled (anti-parallel to the former1-4) in a tetrahedral structure. Fe
3O4 is
used in the water gas shift reaction as a catalyst in the “Haber process”
cubic-spinel-cell contains “8” inter-penetrating oxygens within the tetrahedral sites4,5 and has been occupied through
“1/3” of the Fe atoms, such as diamond structures. The other iron atoms are placed at the octahedral situations with closest atoms arranged as string in six various directions5-7 .
Fe3O4 structures consist of the cubic-close-packed arrays of oxides which all of the Fe(2+) ions
occupies “1/2” of the octahedral locations and the Fe(3+) splits equally along the other octahedral and
tetrahedral locations1-7.
Cyclo-dextrin is extracted from the degradation5 of starch by Bacillusmacerans5,6,
which for the first one has been isolated in the late 9th century5-8. Its abilities for forming complexes
with various organic molecules were discovered immediately there after6-9. By developing the fields
of Chemistry’s Supramolecular8, its complexation
properties have been tightly studied7-10 .Usage
of cyclo-dextrin and its derivatives are desirable in various areas of chemistry7-10, including the
influencing of the organic molecules8-14. By this study
the catalysis’s properties of Fe3O4 nanoparticles @ Cyclodextrins @ (12, 12) SWCNTs for comparing in the area of chemical synthesizes15-20 have been
investigated.
CNT or carbon nanotubes are representatives of Nano materials. CNT is a cylindrically21 shaped-
carbon-material22 with the Nano metric diameter21-30.
Their shapes which are in the structure of hexagonal-mesh23-28,look like a graphite26-30 and these sheets
have wrapped and their two edges have attached together seamlessly31-35.
Although it is a common-place material using in pencil-leads31-33, their unique structures
causes them for presenting a characteristic which had not observed with any other materials30-35.
SWCNTs is classified33 into (1): single-walled of
carbon-Nano- tubes (CNTs), (2): double-walled Nano- tubes (CNTs), (3): multi walled carbon-Nano- tubes (CNTs) according to the number37 of
the layers in a rolled-graphite36-39.
The important attentions in this area are about the SWCNTs diameters, which are varying among (0.4 – 2) nanometer35-40 while the lengths
are in the order of microns(10-6m)32-40, but SWCNTs
with the lengths in the order of centimeters have also been observed recently41-44.
The extremities of the SWCNTs have been nearest with the lids46 of the graphite sheets45-50.
Computational details
A section of our system for Fe3O4@ Cyclodextrins nanoparticles and Fe3O4@ (12, 12) CNTs have been simulated with QM/MM methods and the optimization were carried out51-55 via the
Monte Carlo approaches. These investigations with differences in the force fields are illustrated through comparing52-56 the energies by the force fields of
AMBER57 and OPLS58. In addition, the software of
Hyper-Chem professional version of 7.01 is used for further calculation.
In the noncovalent interaction of two parts of Fe3O4 and Cyclo-dextrin, the density functional methods such as B3LYP are not suitable for describing the van-der-Waals forces in medium55
ranges interaction. So, the QM/MM such as ONIOM method with three classes of (1): high, (2): medium, and (3): low calculations, has been used in this studies between two parts of Fe3O4 and Cyclo-dextrin.
The ab-initio of DFT methods are used for the model of systems through definition of ONIOM53-56 layers and the various semi-empirical55
methods such as pm6 within pseudo=lanl257 order
Fig.1: Non bonded interaction between Cyclodextrins (alpha) and Fe3O4
and the Pm3MM58 for the second and third layers,
respectively59.
The most general of density-functional-theory are inexpressive to exhibit the correlation56
and exchange57 energies for medium-range of
non-bonded systems correctly. In addition, some of the recent works have exhibited the inexactitude of the medium-ranges exchange energies lead to the large principled error in prognostication of the molecular properties51-53.
Electronic structure calculations and geometry optimization have been performed using the m06-DFT. This Functional theories are based on an repetition solution of the Kohn & Sham
equation53,54 of DFT theory in the plane-wave sets
including a projector-augmented-wave-pseudo potential50-54. The PBE55 (Burke, Ernzerh &Perdew),
XC (exchange/correlation) of the GGA (generalized-gradient-approximation) are also has applied. The calculation of the lattice- constant57 and the atomic
coordinate is made by the minimized the systems for the total energies.
The charges transfer56 of electrostatics
potentials derived57 charges were also estimated
using MKS(Merz-Kollman-Singh) and Chelp or chelpG56,57 . The charges calculation method based
on electrostatic-potentials fitting or MESP are not well befit for remedy of the bigger system.
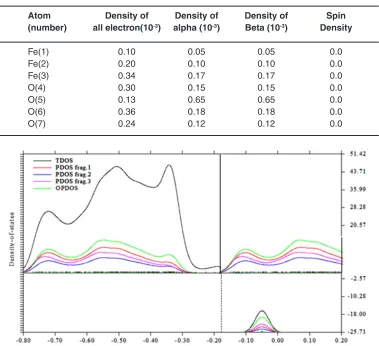
Fig. 2: density of states for Fe3O4@alphaCd@ (12,12)SWCNNTS Table 1: All Electron densities of non-bonded interactions for Fe3O4- Cyclodextrin (a @(12,12)SWCNTs
Atom density of density of density of Spin (number) all electron(10-3) alpha (10-3) beta (10-3) density
Fe(1) 0.10 0.05 0.05 0.0
Fe(2) 0.20 0.10 0.10 0.0
Fe(3) 0.34 0.17 0.17 0.0
O(4) 0.30 0.15 0.15 0.0
O(5) 0.13 0.65 0.65 0.0
O(6) 0.36 0.18 0.18 0.0
By these conditions, changes of the inner-most atomic-charge would not topped toward a remarkable changes of the MESP50-56 for the
molecules, which means that the precise value for the inner-most atomic-charge is not well specified out-side molecules . The agent atomic charge for a molecule might be calculated as the intermediate value over a few molecular conformations50-54.
In an overview of the effect of the basis sets and the “Hamiltonian” on the charges distributions would be found in the references56-58. The charges
densities profile in this kind studies have been extracted from the ûrst principle calculations via an intermediate processing as explained in the references53,54. The interaction energies of these
non-bonded interactions were calculated according to the equation as follows for all items:
Where the “ ” are the stability energies.
Both electron densities of Laplacian and gradient, values of orbital-wave-functions, spin-electron densities, total potential of electrostatics (ESP), electrostatic potential from atomic charges of nucleus, ELF (localization-function for electron ), LOL (locator & orbital-localized), detailed by correlation
hole, as well as the correlation & exchange densities, Becke & Tsirelson, correlation- factor, and the expectation of ionization energies (local) using the Multifunctional55-57 have also been applied in these
kind studies. The contour lines maps were also drawn using the Multi-wfn software55-57. The contour
lines corresponding to the VdW surfaces including of electron densities are defined by Bader and has been drawn in this study55-57. That is clearly useful
for analyzing of distributions for the electrostatics potentials on VdW surfaces. Those contour55 lines
have also been drawn in the gradient-lines55 and
vector-fields-maps56 by the equal option55-57. The
relief62 maps were applied for presenting the height
values at the points. Shaded-surface61 mapswith and
without a projection56 is used in our representation56
of height values at various situation60-62.
RESULT ANd dISCUSSIONS
Monte-Carlo Approaches
The section of ab-initio methods are given by computation that is yielded from principals of theory phenomenon, without inclusion of the experiment information63,64. The important usual
types of ab-initio optimization are called HF, while the primary approximations are called: central-field or CFA. An important method that eschews making the HF problems is popular as the name Quantum-Monte-Carlo*. Also in contrast to the molecular
Table 2: All Electron Energies of non-bonded interactions for Fe3O4- Cyclodextrin (α@(12,12)SWCNTs
Atom Lagrangian kinetic Hamiltonian kinetic Potential energy (number) [G(r)]energy(10-3) [K(r)]energy(10-2) density [U(r)] (10-2)
Fe(1) 0.24 0.45 -0.32
Fe(2) 0.28 0.6 -0.42
Fe(3) 0.12 0.26 -0.60
O(4) 0.26 -0.14 -0.32
O(5) 0.32 -0.20 -0.56
O(6) 0.28 -0.10 -0.22
O(7) 0.10 -0.20 -0.80
dynamics methods which are totally definitive, the Monte Carlo simulation methods are based on using of stochastic significances. On those methods, the systems are included of M atoms which are given in a group of primary orients and interact together. Estimations of those primary configurations are then produced by this consecutive random displacement which works through variation of QMC*, Green’s
functions and diffusion approaches. Those methods work via wave-functions and have evaluated the numerical integrals. Although those optimization might be much more time consuming, these are seems the most precise and suitable ways which are known up to now. An ab-initio calculation gives high quality results and then might yields increasingly high quanty results.
There are 3 levels for accomplishing of any QM/MM optimization in the “Hyper-Chem version 8.0” packages. Firstly sets up the structures of the
molecules with an appropriate starting geometry or coordinates. For the second step it should be chosen suitable optimization including its associated-choices*. For the 3rd step it should be selected a
kind of optimization with the related options. The MC simulation detects a so-called important phase-space* region that is of the lowest energies. Because
of fault of the force fields, these lower energies basin usually (in most cases) does not equal to the normal states, so the rank of native structures produced by the force fields themselves is low-order.
By this work, differences in the force-fields are investigated with comparison the energies optimization using force fields among the Amber, MM+, and OPLS of charmm. In addition, we investigated the polar solvents and the temperature effect (from 260K to 400K) for the stability of single wall of CNTs bonded to CGA or CFA by the various solvent. The QM or quantum mechanics
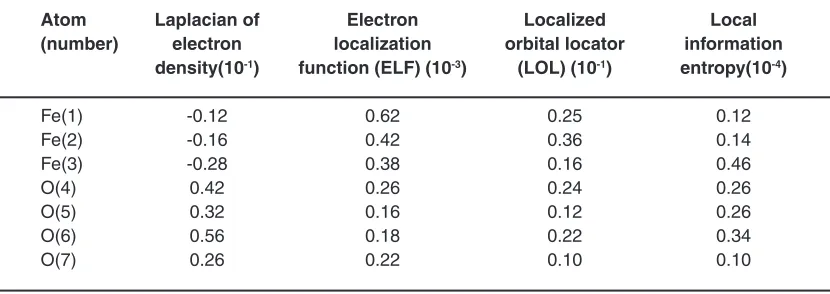
Table 3: Laplacian, ELF, LOL and Local information entropy of non-bonded interactions for Fe3O4- Cyclodextrin ( a @(12,12)SWCNTs
Atom Laplacian of Electron Localized Local (number) electron localization orbital locator information
density(10-1) function (ELF) (10-3) (LOL) (10-1) entropy(10-4)
Fe(1) -0.12 0.62 0.25 0.12
Fe(2) -0.16 0.42 0.36 0.14
Fe(3) -0.28 0.38 0.16 0.46
O(4) 0.42 0.26 0.24 0.26
O(5) 0.32 0.16 0.12 0.26
O(6) 0.56 0.18 0.22 0.34
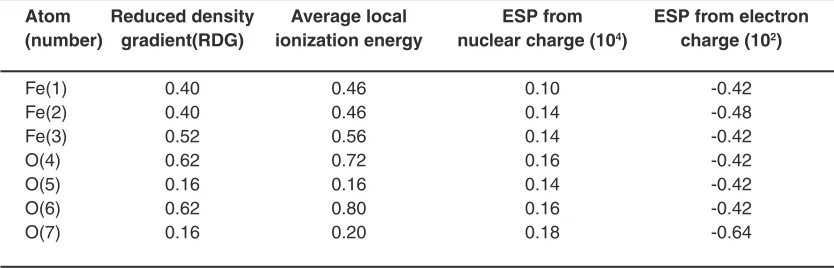
Table4: Average local ionization energy, RdG and ESP of non-bonded interactions for Fe3O4- Cyclodextrin ( a @(12,12)SWCNTs
Atom Reduced density Average local ESP from ESP from electron (number) gradient(RdG) ionization energy nuclear charge (104) charge (102)
Fe(1) 0.40 0.46 0.10 -0.42
Fe(2) 0.40 0.46 0.14 -0.48
Fe(3) 0.52 0.56 0.14 -0.42
O(4) 0.62 0.72 0.16 -0.42
O(5) 0.16 0.16 0.14 -0.42
O(6) 0.62 0.80 0.16 -0.42
O(7) 0.16 0.20 0.18 -0.64
Table 5: Lambada2, Wave function value, Ellipticity of electron density and Eta index of non-bonded interactions for Fe3O4- Cyclodextrin ( a @(12,12)SWCNTs
Atom Lambada2 Wave function Ellipticity of Eta (number) (10-3) value (10-4) electron density index
Fe(1) -0.16 0.42 0.48 -3.41
Fe(2) -0.13 0.88 0.44 -2.54
Fe(3) -0.15 0.67 0.23 0.53
O(4) 0.36 0.29 -0.46 1.41
O(5) 0.11 -0.43 -0.17 0.93
O(6) 0.34 0.33 -0.42 0.38
O(7) 0.23 -0.28 -0.17 0.76
calculation was carried out with the “Hyper-Chem 8.0” program.
This work basically accomplished on the magnetic properties of Fe3O4 in the non-bonding systems of Cyclo-dextrin ( and Fe3O4. The non-bonding interactions are exhibited in figs1- 3. As it is cleared in tables 1-10, the electrical55 property can be
yielded from changing in a non-bonding interaction. Potential energy, electron density, ELF, energy density55, Ellipticity, LOL, eta indexes and ECP of
Fe3O4@Cyclodextrin (@ (12, 12) SWCNTs were calculated of each simulations (Table1-10)55-59.
CONCLUSION
Fe3O4 is Ferro-magnetic including a curie*
temperature of the 858+0.5 K and these
Ferro-magnetism arises for “Fe3O4” because the spin of electrons for the FeIII and FeII in the octahedral location are in a coupled situations together and those spins of the Fe(III) ions in the tetrahedral locations are also coupled together but are in anti-parallel* situations to the previous one. Fe
3O4 is
1. Greenwood, Norman N.; Earnshaw, Alan, Chemistry of the Elements, 1997 (2nd ed.), Butterworth-Heinemann .
2. Ma, Ming; Zhang, Yu; Guo, Zhir4.ui; GU, Ning, Nanoscale Research Letters, 2013, 8 (1), 16
3. Massart, R.; IEEE transactions on magnetics, 17, 2, 1981, 1247–1248
4. Schardinger, F.; Zentralbl. Bakteriol. Abt. II 1911, 29, 188-197; (b) Freudenberg, K.; Schaaf, E. Dumpert, G.; Ploetz, T. Naturwissenschaften, 1937, 27, 850-853
5. Wenz, G. Angew. Chem. Int. Ed. Engl. 1994, 33, 803-822
6. Ueno, A. Supramol. Sci. 1996, 3, 31-36 7. Monajjemi, M. Biophysical Chemistry. 2015
207,114 –127
8. Bagheri,S.; Moosavi,M.S.; Moradiyeh,N.; Z a k e r i , M . ; A t t a r i k h a s r a g h i , N . ; S a g h ay i m a r o u f, N . ; N i y a t z a d e h , G . ; Shekarkhand,M.; Mohammad S. Khalilimofrad, Ahmadin,H.; Ahadi,M.; Molecules 2015, 20, 21636–21657.
9. Monajjemi, M. Theor Chem Acc, 2015, 134:77
10. Monajjemi, M. Journal of Molecular Modeling , 2014, 20, 2507
11. Lee, V.S.; Khaleghian, M.; B. Honarparvar, B.; F. Mollaamin, F. J. Phys.Chem C. 2010, 114, 15315
12. Whittembury G. J Gen Physiol.1960, 43, 43-56
13. Valter, Ström; Richard T.; Olsson, K. V. Rao. J. Mater. Chem, 2010, 20, 4168-4175
14. Mei Fang, Valter Ström, Richard T. Olsson, Lyubov Belova, K. V. Rao, Rapid mixing: Appl. Phys. Lett.2011, 99, 222501
15. Villiers, A. Compt. Rend. 1891, 112, 536-539.
16. Monajjemi, M. Struct Chem. 2012, 23, 551– 580
17. Monajjemi, M.; Boggs, J.E. J. Phys. Chem. A, 2013, 117, 1670 “1684
18. Monajjemi, M.; Khaleghian, M, Journal of Cluster Science. 2011, 22 (4), 673-692 318 19. Monajjemi, M. Chemical Physics. 2013, 425,
29-45
20. Monajjemi, M.; Wayne Jr, Robert. Boggs, J.E. Chemical Physics. 2014, 433, 1-11
21. Mahdavian, L.; Mollaamin, F.; Honarparvar,B.; Fullerenes, Nanotubes and Carbon Nanostructures,2010, 18(1), 45-55, 2010 22. Farmer RE.; Macey RI. BiochiBiophys Acta,
1970, 196, 53-65.
23. Sidel,VW. Solomon AK. J Gen Physiol .1957, 41, 243-257.
24. Monajjemi, M. Falahati, M.; Mollaamin, F.; Ionics, 2013, 19, 155–164
25. Tahan, A.; Mollaamin, F.; Russian Journal of Physical Chemistry A, 2009, 83 (4), 587-597
26. Ketabi, S.; Amiri, A. Russian Journal of Physical Chemistry, 2006, 80(1), S55-S62 27. Monajjemi, M.; Baheri, H.; Mollaamin, F.
Journal of Structural Chemistry.2011 52(1), 54-59
28. X. Blasé.; A. Rubio.; S.G. Louie.; M.L. Cohen.; Europhys. Lett, 1994.28, 335.
29. N.G. Chopra.; A. Zettl.; Solid State Commun 1998, 105, 297.
30. Monajjemi, M.; Heshmat, M.; Haeri, HH, Biochemistry (Moscow), 2006, 71 (1), S113-S122
31. Honarparvar, B.; Nasseri, S. M. .; Khaleghian M. Journal of Structural Chemistry. 2009, 50, 1, 67-77
32. Monajjemi, M.;Honarparvar, B. H. ; Haeri, H. ; Heshmat ,M.; Russian Journal of Physical Chemistry C. 2006, 80(1):S40-S44
33. Mollaamin, F.; Gharibe, S.; Int. J. Phy. Sci, 2011, 6, 1496-1500
34. Mollaamin, F.; Najafpour, J.; Ghadami, S.; Ilkhani, A. R.; Akrami, M. S,. Journal of Computational and Theoretical Nanoscience. 2014, 11 (5), 1290-1298
35. Monajjemi, M.; Mollaamin, F. Journal of Cluster Science, 2012, 23(2), 259-272 36. Monajjemi, M.; Khaleghian, M.; Mollaamin,
F. Molecular Simulation. 2010, 36, 11, 865– 870.
37. Mahdavian, L.; Mangkorntong, N. Fullerenes, Nanotubes and Carbon Nanostructures, 2009, 17 (5), 484-495
38. J. Cumings.; A. Zettl, Solid State Commun.2004, 129, 661
39. R. Ma.; Y. Bando.; H. Zhu.; T. Sato, C. Xu, D. Wu, J. Am.Chem. Soc.2002, 124, 7672, 40. Afsharnezhad, S, Jaafari, M.R..; Mirdamadi,
S..; Mollaamin, F..; Monajemi, H. Chemistry .2008, 17 (1), 55-69
41. Mollaamin, F.; Monajjemi, M. Physics and Chemistry of Liquids .2012, 50, 5, 2012, 596–604
42. Chahkandi B, Zare K, Amiri A. Biochemistry (Mosc). 2005 , 70(3):366-76.
43. Lee, V.S.; Nimmanpipug, P.; Mollaamin, F.; Kungwan, N.; Thanasanvorakun, S.; Russian Journal of Physical Chemistry A, 2009, 83, 13, 2288–2296
44. Jalilian,H.; Monajjemi, M. Japanese Journal of Applied Physics. 2015, 54, 8, 08510 45. Naderi, F.; Mollaamin, F.; Khaleghian, M. J.
Mex. Chem. Soc. 2012, 56(2), 207-211 46. Grimme, S. Angew Chem Int Ed, 2006, (45),
4460–4464, DOI: 10.1002/anie.200600448. 47. Schreiner, P.R.; Fokin, A. A.; Pascal, R.
A Jr.; de Meijere, A. Org. Lett, 2006, (8): 3635–3638.
48. Zhao, Y.; Truhlar, D.G. Org. Lett, 2006, (8):5753–5755.
49. Kohn, W.; Sham, LJ. Phys. Rev, 1965, (140) A: 1133-1138.
50. Perdew, J .P. Burke, K.; Phys. Rev. Lett.1996, (77): 3865-3868.
51. Besler, B.H.; Merz, K.M.; Kollman, P.A. J. comp. Chem, 1990, (11): 431-439,
52. Chirlian, L.E.; Francl, M.M. J.comp.chem, 1987, (8): 894-905, DOI: 10.1002/jcc.540080616 53. Brneman, G.M, Wiberg, K.B. J. Comp Chem,
1990, (11): 361
54. Balderchi, A.; Baroni, S.; Resta, R. Phys. Rev. Lett, 1998, (61): 173.
55. Lu, T.; Chen, F. Acta Chim. Sinica, 2011, 69, 2393-2406.
56. Lu, T.; Chen, F J. Mol. Graph. Model, 2012, (38): 314-323.
57. Lu, T.; Chen F. J. Comp. Chem. 2012, (33) 580-592.
58. Naghsh,F, oriental journal of chemistry, 2015, 31(1).,465-478