Evolution and Dynamics of the Human Gut Virome
Full text
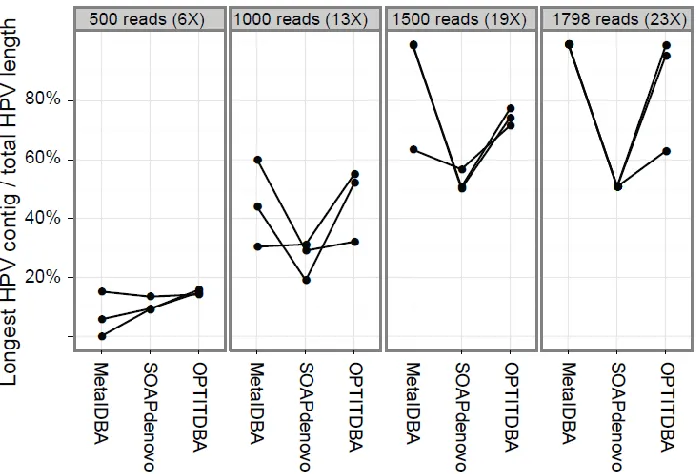
Figure
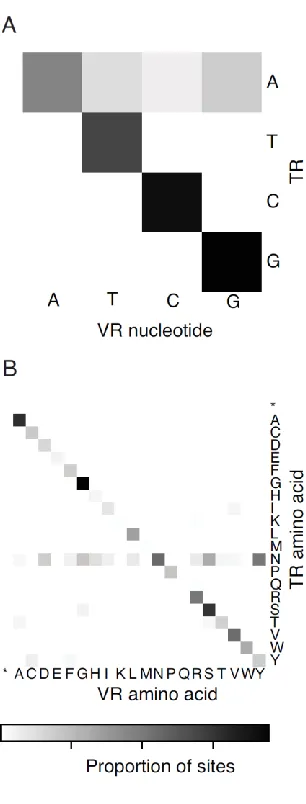
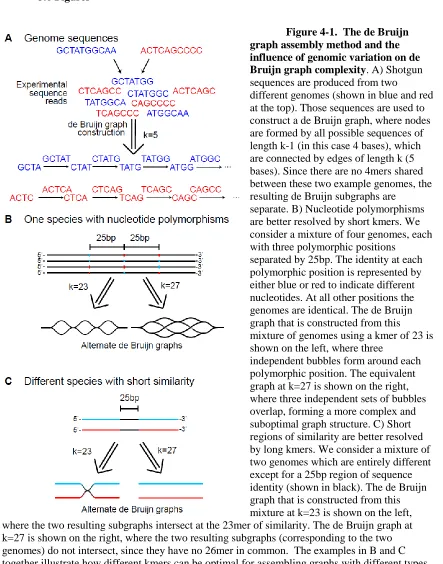
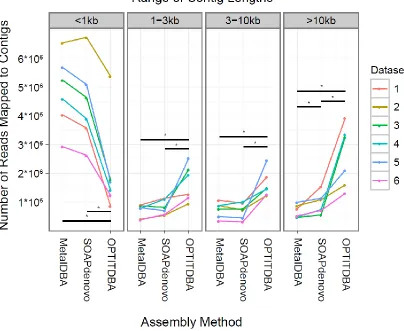
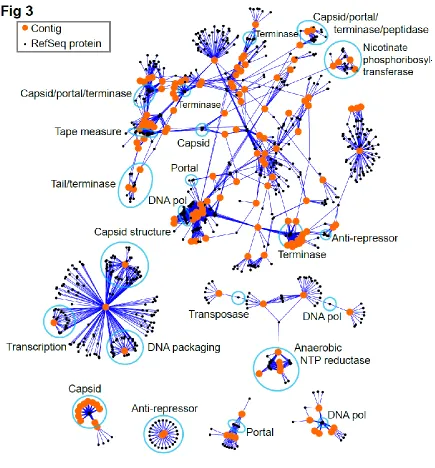

Related documents
In addition, the findings further reveal that none of the studies investigate the mediating effect of the nature of innovation (incremental and radical) on the relationship
The optimized MWPA was concentrated and extracted using ethyl acetate and was further determined for its antimicrobial properties against Gram positive and Gram negative bacterial
There are infinitely many principles of justice (conclusion). 24 “These, Socrates, said Parmenides, are a few, and only a few of the difficulties in which we are involved if
The ethno botanical efficacy of various parts like leaf, fruit, stem, flower and root of ethanol and ethyl acetate extracts against various clinically
Applications of Fourier transform ion cyclotron resonance (FT-ICR) and orbitrap based high resolution mass spectrometry in metabolomics and lipidomics. LC–MS-based holistic metabolic
The following diagram shows just one of many possible configurations when a CP-1650 in Bridge Mode is connected to an RTI audio distribution system (the AD-8 audio
How Many Breeding Females are Needed to Produce 40 Male Homozygotes per Week Using a Heterozygous Female x Heterozygous Male Breeding Scheme With 15% Non-Productive Breeders.
This work looks at the application of one-dimensional diffusion-edited ¹H NMR spectroscopy (1D DOSY) and ¹H NMR with suppression of the ethanol and water signals to the
