EXPERIENCE & REASON
A Novel Monocarboxylate Transporter 8 Gene
Mutation as a Cause of Severe Neonatal Hypotonia
and Developmental Delay
Anastasios Papadimitriou, MDa, Alexandra Mihaela Dumitrescu, MD, PhDb, Antigone Papavasiliou, MDc, Andreas Fretzayas, MDa,
Polyxeni Nicolaidou, MDa, Samuel Refetoff, MDd,e
aThird Department of Pediatrics, University of Athens School of Medicine, Attikon University Hospital, Athens, Greece; Departments ofbMedicine anddPediatrics, eCommittees on Genetics and Molecular Medicine, and J. P. Kennedy Mental Retardation and Developmental Disabilities Center, University of Chicago, Chicago, Illinois; cNeurology Department, Penteli Children’s Hospital, Athens, Greece
The authors have indicated they have no financial relationships relevant to this article to disclose.
ABSTRACT
Monocarboxylate transporter 8 acts as a specific cell membrane transporter for thyroxine and especially triiodothy-ronine into target cells. It is expressed in brain neurons and in many other tissues. The monocarboxylate transporter 8 gene resides on chromosome Xq13.2. An 11-month-old male infant was referred because of severe hypotonia from early life and global developmental delay. Thyroid-function tests showed normal thyrotropin levels and the char-acteristic for the disorder, including high serum triiodothyronine and low thyroxine concentrations. Molecular analysis of the monocarboxylate transporter 8 gene showed that the patient was hemizygous for a novel missense mutation P537L. This case highlights the importance of determining thyroid hormone levels, especially triiodothy-ronine, in infants with severe neonatal hypotonia.
T
HYROID HORMONE (TH)is crucial for the maturation and function of the central nervous system (CNS). TH also exerts a broad range of effects on growth, metabolism, and physiologic function of virtually all tissues.1Most TH effects are mediated by specific THreceptors, which modulate the level of transcription of target genes. Some effects take place in the cytoplasm and are broadly known as nongenomic.2TH is
synthe-sized and secreted from the thyroid gland mainly as the prohormone thyroxine (T4). The principal
bioac-tive form of TH, 3,3⬘,5-triiodothyronine (T3), is
pro-duced in virtually all tissues by outer ring deiodination of thyroxine, a reaction catalyzed by type 1 and type 2 5⬘-deiodinases (D1 and D2, respectively). D1 is found in peripheral tissues, such as the liver, kidney, and thyroid, and is responsible for the conversion of the majority of thyroxine to T3 in the circulation. D2 is
found in the brain, pituitary, thyroid, and skeletal muscle. Thus, D2 has the important function to pro-vide T3to the brain and pituitary.3Type 3 5-deiodinase
converts T4 to the inactive metabolite 3,3⬘,5⬘
-tri-iododothyronine (rT3) and T3to 3,3⬘-diiodothyronine
by inner ring deiodination.4 Type 3 5-deiodinase is
expressed in the brain, especially during fetal devel-opment, as well as in other fetal tissues, the placenta, and the pregnant uterus. For both genomic and non-genomic mechanisms of action, TH has to cross the cell
specific transport proteins, several of which have been identified recently.5
Monocarboxylate transporter 8 (MCT8) acts as a spe-cific transporter of T4and especially for T3.6The gene for
MCT8 resides on chromosome Xq13.2. MCT8 is ex-pressed in brain neurons and other tissues, for example, muscle, bone, and liver. Mutations of the MCT8 gene have only recently been identified; they result in severe psychomotor retardation and abnormal thyroid-func-tion tests (TFTs) that are characterized by a high serum total and free T3and low total and free T4, as well as
low rT3.7
CASE REPORT
An 11-month-old boy was referred because of severe hypotonia from early life and global developmental
de-Key Words:monocarboxylate transporter 8, neonatal hypotonia, developmental delay, thyroid hormones
Abbreviations:TH—thyroid hormone; CNS— central nervous system; T3—3,3⬘,5-triiodothyronine; D1—type 1 5⬘-deiodinase; D2—type 2 5⬘-deiodinase;
T4—thyroxine; rT3—3,3⬘,5⬘-triiododothyronine; MCT8 —monocarboxylate transporter
8; TFT—thyroid-function test; L-T4—L-thyroxine
www.pediatrics.org/cgi/doi/10.1542/peds.2007-1247 doi:10.1542/peds.2007-1247
Accepted for publication May 24, 2007
Address correspondence to Anastasios Papadimitriou, MD, Pediatric Endocrinology Unit, Third Department of Pediatrics, University of Athens School of Medicine, Attikon University Hospital, Rimini 1, Haidari, Athens 124 62, Greece. E-mail: anpapad@med.uoa.gr
lay. He was born after an uncomplicated term pregnancy with a birth weight of 2.5 kg. His parents were not consanguineous. No perinatal distress was reported, but he was noted to have a weak cry. At presentation his body length was 77 cm (⬃80th percentile), weight was 8.1 kg (10th percentile), and head circumference was 46 cm (55th percentile). He exhibited severe generalized hypotonia and decreased muscle strength, hyperactive deep tendon reflexes, and severe head lag and was un-able to sit independently. He showed no signs of thyroid dysfunction, and the thyroid was nonpalpable. His car-diovascular examination was normal. Neonatal thyroid screening by measurement of thyrotropin was within the reference range (ie, spot thyrotropin⬍25⌱U/mL). Biochemical investigation included serum glucose, se-rum urea nitrogen, sese-rum creatinine, K⫹, Na⫹, Cl⫺, Ca⫹2, phosphorus, alkaline phosphatase, Mg⫹2,
aspar-tate aminotransferase, alanine aminotransferase,␥
-glu-tamyl transpeptidase, lactate dehydrogenase, total cholesterol, low-density lipoprotein cholesterol, high-density lipoprotein cholesterol, triglycerides, lactate, and total protein, all of which were within the reference ranges except for a mildly elevated lactate level of 23.9 mg/dL (reference:⬍20 mg/dL). At 11 months of age, the serum total and free T3levels were high, T4was low, and
thyrotropin was within the reference range (Table 1). A brain MRI showed decreased myelination of the subcor-tical tissue and thalamus. Clinical presentation, bio-chemical profile (high T3and low T4), brain MRI, and
the gender of the patient suggested the possibility of an
MCT8gene mutation. Family history was also suggestive of an X-linked disorder (Fig 1). His maternal uncle pre-sented an unidentified neurologic disorder preceding death at the age of 8 years. The patient had 3 brothers, 2 of them without a neurologic disorder; however, the third one had presented muscular hypotonia since birth and died at the age of 9 months from aspiration pneu-monia. Molecular analysis of theMCT8gene of the fam-ily (Fig 1) showed that the patient and his mother, maternal aunt, and grandmother carried a point muta-tion, a single nucleotide substitution (cytosine to thymi-dine) in exon 5, which results in the replacement of the normal proline 537 with a leucine. This mutation, P537L, has not been reported previously. L-thyroxine (L-T4) administration (37.5 g/day) resulted at 13
months (Table 1) in the restoration of a normal free T4
level of 1.1 ng/dL, a further increase in the free T3level
to 8.5 pg/mL, and a suppressed thyrotropin level of 0.19
⌱U/mL but did not improve the patient’s neurologic condition. The TFTs at 18 months while on 25g of L-T4
daily (Table 1 and Fig 1) showed an increased total T3
concentration of 280 ng/dL with a normal T4level of 8.3 g/dL and a normal rT3level of 19.9 ng/dL, likely be-TABLE 1 TFTs of the Propositus With Age and With L-T4treatment
Treatment Reference
Range
Patient Age
11 mo, No Treatment
13 mo, L-T4at
37.5g/d
18 mo, L-T4at
25g/da
TT4,g/dL 7.2–14.9 5.48 NA 8.30
FT4, ng/dL 0.8–2.0 0.52 1.1 NA
TT3, ng/dL 105–245 302 NA 280
FT3, pg/mL 2.0–4.0 4.26 8.50 NA
Thyrotropin,U/mL 0.4–5.0 2.64 0.19 0.90
rT3, ng/dL 15–35 NA NA 19.9
TG, ng/mL 1–35 NA NA 6.0
Ab TG/TPO, IU/mL NA NA NA Negative
NA indicates not applicable; TT4, total T4; FT4, free T4; TT3, total T3; FT3, free T3; TG, thyroglobulin;
Ab TG/TPO, thyroglobulin/thyroid peroxidase antibodies.
aFor reference values of TFT at 18 months, see Fig 1.
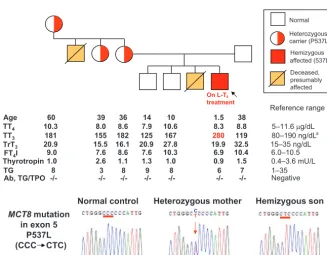
Heterozygous mother Hemizygous son
Normal control
C T
Age 60 39 36 14 10 1.5 38
TT4 10.3 8.0 8.6 7.9 10.6 8.3 8.8 5–11.6µg/dL
TT3 181 155 182 125 167 280 119 80–190 ng/dL
TrT3 20.9 15.5 16.1 20.9 27.8 19.9 32.5 15–35 ng/dL
FT4I 9.0 7.6 8.6 7.6 10.3 6.9 10.4 6.0–10.5
1.0 2.6 1.1 1.3 1.0 0.9 1.5
TG 8 3 8 9 8 6 7 1–35
Ab, TG/TPO -/- -/- -/- -/- -/- -/-
-/-0.4–3.6 mU/L
Negative Reference range
On L-T treatment
MCT8 mutation in exon 5
P537L (CCC CTC)
Heterozygous affected (537L) Deceased, presumably affected Normal carrier (P537L) Hemizygous a Thyrotropin 4 FIGURE 1
Pedigree, TFTs, and theMCT8gene mutation (P537L) in members of the family. The propositus is indicated by a black arrow. TFT values are aligned under each subject symbol. TT4indicates total T4; TT3, total T3; TrT3, total
reverse T3; TG, thyroglobulin; FT4I, free T4index. High
values are shown in red type. Note that the TFTs of the propositus were measured in a sample obtained while receiving 25g of L-T4 perday.aThe reference range for
children is 90 to 210 ng/dL. Electropherograms show part of theMCT8exon 5 in a normal control, the carrier mother, and the propositus. The red arrow indicates the location of the mutation, and the correspondent codon is underlined.
e200 PAPADIMITRIOU et al
at Viet Nam:AAP Sponsored on August 29, 2020
www.aappublications.org/news
cause of normalization of the circulating T4level. TFTs of
the 3 heterozygous female carriers and 3 unaffected family members were within the reference range (Fig 1). However, serum T3concentrations in the carrier females
tended to be higher and those of rT3lower relative to the
unaffected family members. More specifically, the T3
level was 181, 155, and 182 ng/dL in the heterozygotes compared with 125, 167, and 119 ng/dL in the unaf-fected. Similarly, the rT3level was 20.9, 15.5, and 16.1
ng/dL in the heterozygotes compared with 20.9, 27.8, and 32.5 ng/dL in the unaffected subjects (Fig 1). This trend of intermediate values in heterozygous females, though noted previously, and its physiologic mechanism confirmed7,8 has no diagnostic value in predicting the
carrier state of individual subjects.
DISCUSSION
Loss-of-function mutations of theMCT8gene result in a dramatic reduction in T3uptake by the brain neurons,
cerebral hypothyroidism, and a neurologic phenotype of profound hypotonia and developmental delay, described in this and other patients.7,9–14 Evidence for the
impor-tance of MCT8 on brain function in humans became evident with the identification of theMCT8gene muta-tions in 2 boys with neuropsychiatric defects and elevated serum T3 levels.7 This was followed by the
publication describing 5 more unrelated boys9and,
ulti-mately, the realization that patients with Allan-Hern-don-Dudley syndrome also carry a mutation in theMCT8
gene.10 This syndrome was among the first X-linked
mental retardation syndromes to be clinically described. Based on⬎100 subjects, the clinical manifestations are now well defined. In infancy and childhood, the char-acteristics are marked hypotonia, weakness, generalized muscular atrophy, and delay of developmental mile-stones. Head circumference is normal; however, a num-ber of patients present acquired microcephaly usually manifesting after the seventh month of life. Other neu-rologic manifestations include spastic paraplegia with hyperreflexia, clonus, and Babinski reflexes. In adult life, hypotonia turns to spasticity. Cognitive develop-ment is severely impaired.11MCT8 deficiency has been
also associated with X-linked paroxysmal dyskinesia and progressive atrophy of the cerebrum, basal ganglia, and midbrain.15
The recent development of animal models of MCT8 deficiency8,16has helped in unraveling the
pathophysio-logic mechanism that gives rise to the abnormal TFTs. The reduced entry of T4into brain cells stimulates D2
activity, whereas excess of TH in liver, reflecting cellular uptake of serum T3through transporters different from
MCT8, stimulates D1 activity. This results in the forma-tion of more T3 and consumption of T4. The reduced
uptake of T3further enhances T3accumulation in serum.
The low rT3 levels are explained by the increased rT3
metabolism because of increased D1 activity and the consumption of T4. The role of MCT8 on the CNS is less
well understood. It is likely that MCT8 deficiency results in the reduction of T3uptake by brain neurons
produc-ing early CNS hypothyroidism in the embryo resultproduc-ing in
the possibility that MCT8 could transport yet another substrate, vital to CNS function, has not been excluded. In our patient, thyrotropin suppression and normal rT3 levels when serum T4 were normalized, while on
L-T4treatment, suggest a more limited role of MCT8 on
T4 transport into the pituitary thyrotrophs. The
in-creased serum lactate levels are compatible with the concept of peripheral thyrotoxicosis.
Whether tests used for neonatal screening of hypo-thyroidism could detect MCT8 defects is not known, because this condition was not recognized until very recently.7,9 Retrospective review showed that
thyro-tropin measurements had been within the reference range at birth.7,14,17This is not surprising given the broad
reference range at birth and that only a small number of the affected individuals manifest later minimal increases in thyrotropin.7,9 In contrast, blood T
4 was low at the
neonatal screen in both subjects in whom this test was reported.7,17 However, because the T
4 concentration is
often low in neonates because of reduced serum T4
binding, this common trivial finding has led most screen-ing programs to abandon T4 testing in favor of
thyro-tropin. T3values are normally very low at birth and rise
rapidly in the first few days of life. High neonatal T3
values may be of diagnostic value for MCT8 defects, but the precise timing of sampling and prematurity could be seriously confounding.
Currently the best method for early diagnosis is to obtain TFTs in infants with hypotonia and feeding diffi-culties, which should be evident to the astute pediatri-cian by the second week of life. An earlier diagnosis or even prenatal testing should be reserved for infants born to mothers known to be heterozygous for anMCT8gene mutation.
Most vexing is the failure of affected individuals to respond to TH treatment, even when given at an early age. Development of TH analogs that enter brain cells by alternative routes to MCT8 are in progress. Currently, prenatal testing and genetic counseling of carrier women can prevent the transmission of this defect to male off-spring.
CONCLUSIONS
MCT8mutations result in severe neonatal hypotonia and global developmental delay. Moreover, this case high-lights the importance of determining TH levels, espe-cially T3, in patients with early postnatal hypotonia.
ACKNOWLEDGMENT
This work was supported by National Institutes of Health grants DK15070 and RR00055.
REFERENCES
1. Brent GA. The molecular basis of thyroid hormone action. N Engl J Med.1994;331:847– 853
2. Bassett JH, Harvey CB, Williams GR. Mechanisms of thyroid hormone receptor-specific nuclear and extra nuclear actions. Mol Cell Endocrinol.2003;213:1–11
PR. Regional expression of the type 3 iodothyronine deiodinase messenger ribonucleic acid in the rat central nervous system and its regulation by thyroid hormone. Endocrinology. 1999; 140:784 –790
5. Jansen J, Friesema EC, Milici C, Visser TJ. Thyroid hormone transporters in health and disease.Thyroid.2005;15:757–768 6. Friesema EC, Ganguly S, Abdalla A, Manning Fox JE, Halestrap
AP, Visser TJ. Identification of monocarboxylate transporter 8 as a specific thyroid hormone transporter.J Biol Chem.2003; 278:40128 – 40135
7. Dumitrescu AM, Liao XH, Best TB, Brockmann K, Refetoff S. A novel syndrome combining thyroid and neurological abnor-malities is associated with mutations in a monocarboxylate transporter gene.Am J Hum Genet.2004;74:168 –175 8. Dumitrescu AM, Liao XH, Weiss RE, Millen K, Refetoff S.
Tissue-specific thyroid hormone deprivation and excess in monocarboxylate transporter (mct) 8-deficient mice. Endocri-nology.2006;147:4036 – 4043
9. Friesema EC, Grueters A, Biebermann H, et al. Association between mutations in a thyroid hormone transporter and se-vere X-linked psychomotor retardation. Lancet. 2004;364: 1435–1437
10. Schwartz CE, May MM, Carpenter NJ, et al. Allan-Herndon-Dudley syndrome and the monocarboxylate transporter 8 (MCT8) gene.Am J Hum Genet.2005;77:41–53
11. Maranduba CMC, Friesema ECH, Kok F, et al. Decreased cel-lular uptake and metabolism in Allan-Herndon-Dudley syn-drome (AHDS) due to a novel mutation in the MCT8 thyroid hormone transporter.J Med Genet.2006;43:457– 460
12. Holden KR, Zuniga OF, May MM, et al. X-linked MCT8 gene mutations: characterization of the pediatric neurologic pheno-type.J Child Neurol.2005;20:852– 857
13. Herzovich V, Vaiani E, Marino R, et al. Unexpected peripheral markers of thyroid function in a patient with a novel mutation of the MCT8 thyroid hormone transporter gene. Horm Res. 2006;67:1– 6
14. Kakinuma H, Itoh M, Takahashi H. A novel mutation in the monocarboxylate transporter 8 gene in a boy with putamen lesions and low free T4 levels in cerebrospinal fluid.J Pediatr. 2005;147:552–554
15. Brockmann K, Dumitrescu AM, Best TT, Hanefeld F, Refetoff S. X-linked paroxysmal dyskinesia and severe global retardation caused by defective MCT8 gene.J Neurol.2005;252:663– 666 16. Trajkovic M, Visser TJ, Mittag J, et al. Abnormal thyroid
hor-mone metabolism in mice lacking the monocarboxylate trans-porter 8.J Clin Invest.2007;117:627– 635
17. Visser WE, Friesema EC, Jansen J, et al. Large X-chromosome deletion affecting the MCT8 gene in a severely retarded boy with elevated serum T3[abstract].Thyroid.2006;16:1072.
e202 PAPADIMITRIOU et al
at Viet Nam:AAP Sponsored on August 29, 2020
www.aappublications.org/news
DOI: 10.1542/peds.2007-1247
2008;121;e199
Pediatrics
Andreas Fretzayas, Polyxeni Nicolaidou and Samuel Refetoff
Anastasios Papadimitriou, Alexandra Mihaela Dumitrescu, Antigone Papavasiliou,
Neonatal Hypotonia and Developmental Delay
A Novel Monocarboxylate Transporter 8 Gene Mutation as a Cause of Severe
Services
Updated Information &
http://pediatrics.aappublications.org/content/121/1/e199 including high resolution figures, can be found at:
References
http://pediatrics.aappublications.org/content/121/1/e199#BIBL This article cites 17 articles, 2 of which you can access for free at:
Subspecialty Collections
http://www.aappublications.org/cgi/collection/genetics_sub
Genetics
following collection(s):
This article, along with others on similar topics, appears in the
Permissions & Licensing
http://www.aappublications.org/site/misc/Permissions.xhtml in its entirety can be found online at:
Information about reproducing this article in parts (figures, tables) or
Reprints
DOI: 10.1542/peds.2007-1247
2008;121;e199
Pediatrics
Andreas Fretzayas, Polyxeni Nicolaidou and Samuel Refetoff
Anastasios Papadimitriou, Alexandra Mihaela Dumitrescu, Antigone Papavasiliou,
Neonatal Hypotonia and Developmental Delay
A Novel Monocarboxylate Transporter 8 Gene Mutation as a Cause of Severe
http://pediatrics.aappublications.org/content/121/1/e199
located on the World Wide Web at:
The online version of this article, along with updated information and services, is
by the American Academy of Pediatrics. All rights reserved. Print ISSN: 1073-0397.
the American Academy of Pediatrics, 345 Park Avenue, Itasca, Illinois, 60143. Copyright © 2008 has been published continuously since 1948. Pediatrics is owned, published, and trademarked by Pediatrics is the official journal of the American Academy of Pediatrics. A monthly publication, it
at Viet Nam:AAP Sponsored on August 29, 2020
www.aappublications.org/news