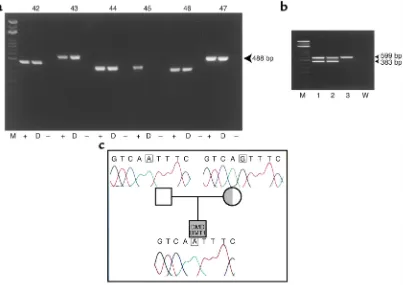
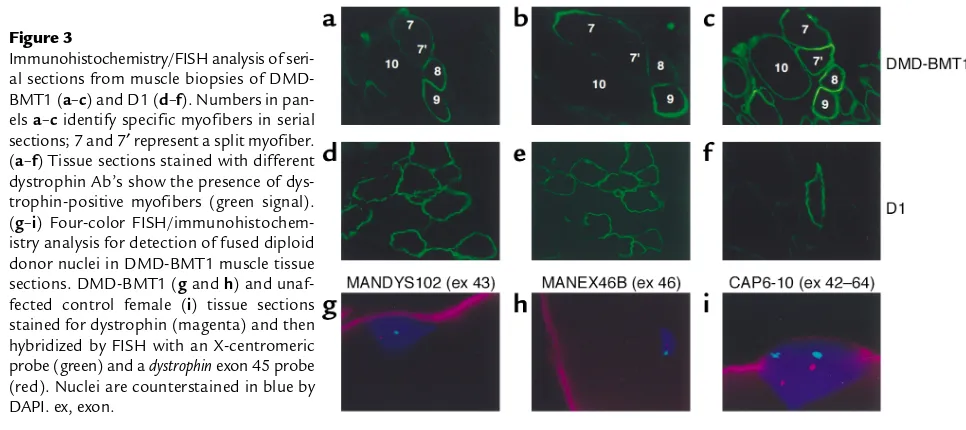
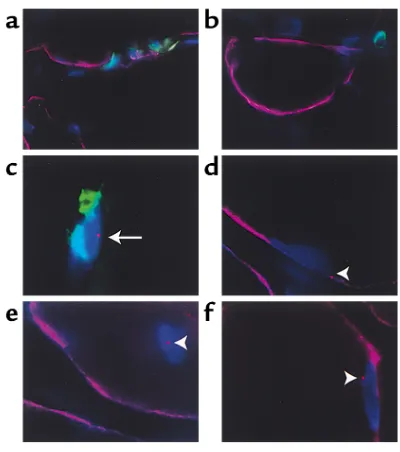
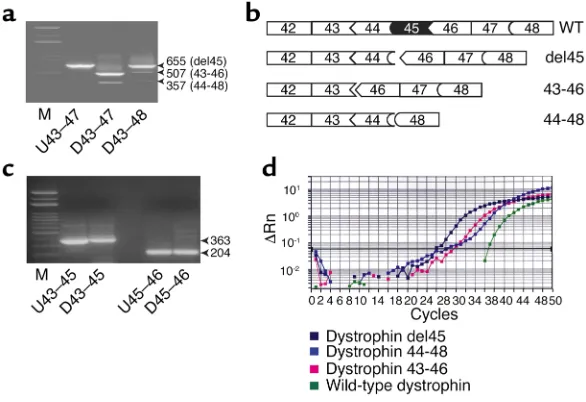
Long term persistence of donor nuclei in a Duchenne muscular dystrophy patient receiving bone marrow transplantation
Full text
Figure
Related documents
METHODS: In this study 100 patients of COPD coming to OPD as well as indoor cases in NCH, Surat from JANUARY 2016 to NOVEMBER 2016 for correlation of 6 Minutes Walk Test
Though the clearest sources of international law do not explicitly provide the right to free legal assistance for children in removal proceedings, trends in
At the end the introduced assay with primer combination (A) is able to detect the point mutation in all samples of formalin-fixed, paraffin-embedded tissue (FFPE) and blood (see
The aim of this work was to evaluate the prevalence of Campylobacter jejuni (as affected by refrigerated storage) in chicken samples obtained from the wholesale poultry market in
(2004) reported a limited randomised double-blinded, placebo-controlled trial investigating the use of phenytoin for the prevention of early post- traumatic seizures in children
In this study, because there was no association between periodontal disease-causing bacteria and the dietary intervention group, the improvement in peri- odontal disease may be
AAP National Center of Medical Home Initiatives for Children With Special Needs (training programs and materials and other resources for pediatricians including materials to help
For all the tested operating conditions, the effect of CNG and diesel fuel injection pressure, together with the amount of fuel injected during the pilot injection, were analyzed
