Acta Cryst.(2001). E57, o827±o828 DOI: 10.1107/S1600536801013022 Nie, Xu, Li and Chiang C7H6N2O4
o827
organic papers
Acta Crystallographica Section E Structure Reports Online
ISSN 1600-5368
2,6-Dinitrotoluene
Jing-Jing Nie,aDuan-Jun Xu,a*
Zhen-Yu Liaand Michael Y.
Chiangb
aDepartment of Chemistry, Zhejiang University,
Hangzhou, Zhejiang, China, andbDepartment
of Chemistry, National Sun Yat-Sen University, Kaohsiung, Taiwan
Correspondence e-mail: [email protected]
Key indicators
Single-crystal X-ray study T= 293 K
Mean(C±C) = 0.004 AÊ Rfactor = 0.046 wRfactor = 0.133 Data-to-parameter ratio = 8.9
For details of how these key indicators were automatically derived from the article, see http://journals.iucr.org/e.
#2001 International Union of Crystallography Printed in Great Britain ± all rights reserved
Crystals of 2,6-dinitrotoluene, C7H6N2O4, were obtained from
a methanol solution of the mixture of 2,6- and 2,4-dinitro-toluene. The P212121 space group shows the compound is
chiral, but the absolute con®guration was not determined reliably. The nitro group planes are inclined to the benzene plane with dihedral angles of 53.1 (1) and 38.1 (1),
respec-tively. The repulsion between methyl and nitro groups results
in a rather small CÐCÐC angle of 112.6 (2) within the
benzene ring.
Experimental
Single crystals of the title chiral compound were obtained from a methanol solution containing 2,6- and 2,4-dinitrotoluene when we tried to separate the isomers.
Crystal data C7H6N2O4
Mr= 182.14
Orthorhombic,P212121
a= 7.830 (2) AÊ
b= 13.683 (3) AÊ
c= 7.296 (2) AÊ
V= 781.7 (3) AÊ3
Z= 4
Dx= 1.548 Mg mÿ3
Dmnot measured
MoKradiation Cell parameters from 20
re¯ections
= 11.7±21.5
= 0.13 mmÿ1
T= 293 (2) K Prismatic, yellow 0.300.240.20 mm Data collection
Rigaku AFC-7Rdiffractometer
!/2scans
Absorption correction: scan (Northet al., 1968)
Tmin= 0.963,Tmax= 0.974
1061 measured re¯ections 1061 independent re¯ections 906 re¯ections withI> 2(I)
max= 27.5
h= 0!10
k= 0!17
l= 0!9
3 standard re¯ections every 100 re¯ections intensity decay: 0.3%
Re®nement Re®nement onF2
R[F2> 2(F2)] = 0.046
wR(F2) = 0.133
S= 1.05 1061 re¯ections 119 parameters
H-atom parameters constrained
w= 1/[2(F
o2) + (0.0748P)2
+ 0.2426P]
whereP= (Fo2+ 2Fc2)/3
(/)max< 0.001
max= 0.27 e AÊÿ3
min=ÿ0.23 e AÊÿ3
Extinction correction:SHELXL93 Extinction coef®cient: 0.0719 (109)
Table 1
Selected geometric parameters (AÊ,).
O1ÐN1 1.216 (3) O2ÐN1 1.213 (4) O3ÐN2 1.221 (3)
O4ÐN2 1.206 (4) N1ÐC2 1.482 (3) N2ÐC6 1.476 (3)
O1ÐN1ÐO2 124.8 (2) O1ÐN1ÐC2 117.5 (3) O2ÐN1ÐC2 117.6 (3) O4ÐN2ÐO3 124.0 (3)
O4ÐN2ÐC6 117.4 (3) O3ÐN2ÐC6 118.7 (2) C2ÐC1ÐC6 112.6 (2)
H atoms were located in calculated positions and constrained to ride on their parent C atoms during re®nement. The absolute con®guration could not be reliably determined.
Data collection: MSC/AFC Diffractometer Control Software
(Molecular Structure Corporation, 1985a, 1992a); cell re®nement:
MSC/AFC Diffractometer Control Software; data reduction:
TEXSAN (Molecular Structure Corporation, 1985b, 1992b); program(s) used to solve structure: SHELXS93 (Sheldrick, 1993); program(s) used to re®ne structure:SHELXL93; molecular graphics:
XP(Siemens, 1994).
The project was supported by the National Natural Science Foundation of China (29973036).
References
Molecular Structure Corporation (1985a, 1992a).MSC/AFC Diffractometer Control Software. MSC, 3200 Research Forest Drive, The Woodlands, TX 77381, USA.
Molecular Structure Corporation (1985b, 1992b). TEXSAN. MSC, 3200 Research Forest Drive, The Woodlands, TX 77381, USA.
North, A. C. T., Phillips, D. C. & Mathews, F. S. (1968).Acta Cryst.A24, 351± 359.
Sheldrick, G. M. (1993). SHELXS93 and SHELXL93. University of GoÈttingen, Germany.
Siemens (1994).XP. Version 5.03. Siemens Analytical X-ray Instruments Inc., Madison, Wisconsin, USA.
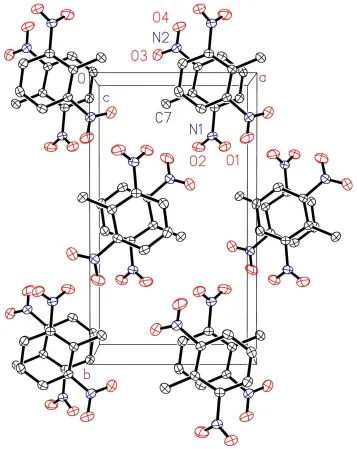
Figure 1
supporting information
sup-1
Acta Cryst. (2001). E57, o827–o828supporting information
Acta Cryst. (2001). E57, o827–o828 [doi:10.1107/S1600536801013022]
2,6-Dinitrotoluene
Jing-Jing Nie, Duan-Jun Xu, Zhen-Yu Li and Michael Y. Chiang
S1. Comment
no comment!
S2. Experimental
Single crystals of the title chiral compound were obtained from a methanol solution containing 2,6- and
2,4-dinitro-toluene when we tried to separate the isomers.
S3. Refinement
H atoms were located in calculated positions and ridden on parent C atoms during refinements. The absolute
Figure 1
View of the crystal packing of the title compound with 30% probability displacement ellipsoids, showing the overlap of
adjacent benzene rings. H atoms have been omitted for clarity.
2,6-Dinitrotoluene
Crystal data
C7H6N2O4 Mr = 182.14
Orthorhombic, P212121 a = 7.830 (2) Å
b = 13.683 (3) Å
c = 7.296 (2) Å
V = 781.7 (3) Å3 Z = 4
F(000) = 376
Dx = 1.548 Mg m−3
Mo Kα radiation, λ = 0.71073 Å Cell parameters from 20 reflections
θ = 11.7–21.5°
supporting information
sup-3
Acta Cryst. (2001). E57, o827–o828Data collection
Rigaku AFC-7R diffractometer
Radiation source: fine-focus sealed tube Graphite monochromator
ω/2θ scans
Absorption correction: psi scan (North et al., 1968)
Tmin = 0.963, Tmax = 0.974 1061 measured reflections
1061 independent reflections 906 reflections with I > 2σ(I)
Rint = 0.000
θmax = 27.5°, θmin = 3.0°
h = 0→10
k = 0→17
l = 0→9
3 standard reflections every 100 reflections intensity decay: 0.3%
Refinement
Refinement on F2
Least-squares matrix: full
R[F2 > 2σ(F2)] = 0.046 wR(F2) = 0.133 S = 1.05 1061 reflections 119 parameters 0 restraints
Primary atom site location: structure-invariant direct methods
Secondary atom site location: difference Fourier map
Hydrogen site location: inferred from neighbouring sites
H-atom parameters constrained
w = 1/[σ2(F
o2) + (0.0748P)2 + 0.2426P]
where P = (Fo2 + 2Fc2)/3
(Δ/σ)max < 0.001
Δρmax = 0.27 e Å−3
Δρmin = −0.23 e Å−3
Extinction correction: SHELXL93, Fc*=kFc[1+0.001xFc2λ3/sin(2θ)]-1/4
Extinction coefficient: 0.0719 (109)
Special details
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes)
are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes.
Refinement. Refinement on F2 for ALL reflections except for 0 with very negative F2 or flagged by the user for potential
systematic errors. Weighted R-factors wR and all goodnesses of fit S are based on F2, conventional R-factors R are based
on F, with F set to zero for negative F2. R-factors based on F2 are statistically about twice as large as those based on F,
and R- factors based on ALL data will be even larger.
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
x y z Uiso*/Ueq
H5 0.8401 (4) −0.1759 (2) 0.9188 (5) 0.057* C6 0.6635 (3) −0.0686 (2) 0.9191 (4) 0.0400 (6) C7 0.4516 (4) 0.0738 (2) 0.8999 (6) 0.0528 (8) H7A 0.3695 (4) 0.0218 (2) 0.9067 (6) 0.079* H7B 0.4392 (4) 0.1075 (2) 0.7853 (6) 0.079* H7C 0.4330 (4) 0.1187 (2) 0.9990 (6) 0.079*
Atomic displacement parameters (Å2)
U11 U22 U33 U12 U13 U23
O1 0.0618 (13) 0.0413 (10) 0.089 (2) −0.0103 (11) −0.0006 (14) 0.0130 (12) O2 0.078 (2) 0.0411 (12) 0.094 (2) 0.0051 (12) 0.013 (2) −0.0150 (12) O3 0.0462 (12) 0.0585 (13) 0.095 (2) −0.0100 (10) 0.0075 (13) 0.0034 (14) O4 0.078 (2) 0.0458 (12) 0.130 (3) −0.0177 (13) 0.001 (2) −0.027 (2) N1 0.0455 (12) 0.0305 (10) 0.070 (2) −0.0027 (9) −0.0062 (14) 0.0001 (12) N2 0.0430 (12) 0.0389 (11) 0.069 (2) −0.0069 (10) −0.0044 (13) 0.0003 (13) C1 0.0363 (12) 0.0333 (12) 0.0489 (13) 0.0032 (10) 0.0002 (13) 0.0006 (12) C2 0.0407 (13) 0.0298 (11) 0.053 (2) −0.0001 (10) 0.0006 (15) −0.0022 (12) C3 0.0378 (13) 0.0425 (14) 0.062 (2) −0.0053 (11) −0.004 (2) 0.0046 (15) C4 0.0345 (12) 0.0471 (14) 0.065 (2) 0.0072 (11) 0.000 (2) 0.0036 (15) C5 0.0461 (14) 0.0340 (12) 0.061 (2) 0.0068 (11) −0.001 (2) −0.0013 (13) C6 0.0379 (12) 0.0299 (11) 0.0523 (15) −0.0041 (10) −0.0014 (14) −0.0009 (12) C7 0.0397 (13) 0.0395 (13) 0.079 (2) 0.0081 (12) 0.002 (2) 0.005 (2)
Geometric parameters (Å, º)
O1—N1 1.216 (3) C3—C4 1.386 (4)
O2—N1 1.213 (4) C3—H3 0.93
O3—N2 1.221 (3) C4—C5 1.372 (4)
O4—N2 1.206 (4) C4—H4 0.93
N1—C2 1.482 (3) C5—C6 1.382 (4)
N2—C6 1.476 (3) C5—H5 0.93
C1—C2 1.389 (4) C7—H7A 0.96
C1—C6 1.404 (3) C7—H7B 0.96
C1—C7 1.505 (4) C7—H7C 0.96
C2—C3 1.380 (4)
O1—N1—O2 124.8 (2) C5—C4—C3 119.5 (3)
O1—N1—C2 117.5 (3) C5—C4—H4 120.3
O2—N1—C2 117.6 (3) C3—C4—H4 120.3
O4—N2—O3 124.0 (3) C4—C5—C6 119.5 (2)
O4—N2—C6 117.4 (3) C4—C5—H5 120.2
O3—N2—C6 118.7 (2) C6—C5—H5 120.24
C2—C1—C6 112.6 (2) C5—C6—C1 124.3 (2)
C2—C1—C7 123.7 (2) C5—C6—N2 115.7 (2)
C6—C1—C7 123.5 (2) C1—C6—N2 120.0 (2)
C3—C2—C1 125.4 (2) C1—C7—H7A 109.47
supporting information
sup-5
Acta Cryst. (2001). E57, o827–o828C1—C2—N1 118.8 (2) H7A—C7—H7B 109.5
C2—C3—C4 118.6 (3) C1—C7—H7C 109.5
C2—C3—H3 120.7 H7A—C7—H7C 109.5