TGFbeta induces apoptosis and EMT in primary mouse hepatocytes independently of p53, p21(Cip1) or Rb status
Full text
Figure
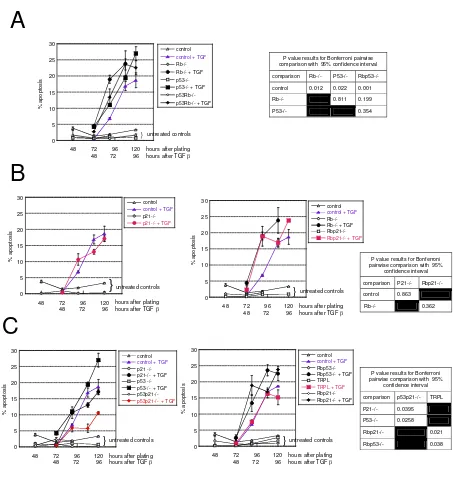
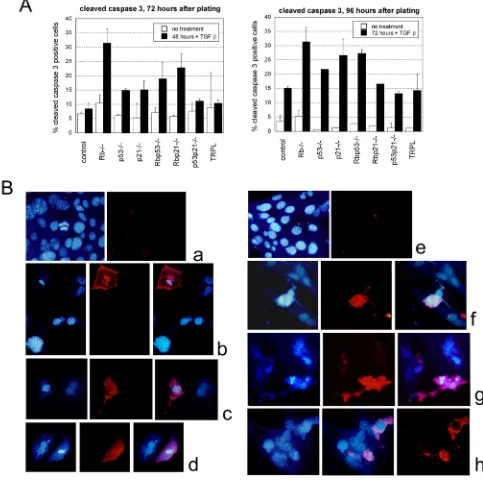
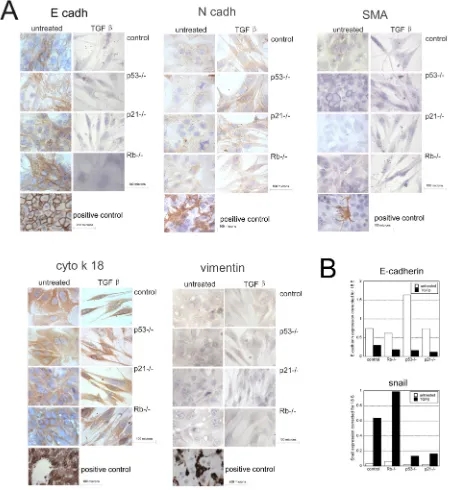
Related documents
In sum, we need to help children to successfully develop social- emotional competencies and mental health wellbeing, reduce behavioural problems, increase desirable
El caso argentino ilustra cómo impactan en el perio dismo los límites al acceso a las fuentes oficiales y a la información pública impuestos desde la oficina de prensa de
Conf: Confidence; EPM: Emerging pattern mining; EPs: Emerging patterns; FPR: False positive rate; G-mean: Geometric mean of true positive rate and true negative rate; GR: Growth
matrices of the multivariate time series data of solar events as adjacency matrices of labeled graphs, and applying thresholds on edge weights can model the solar flare
The Houston (First) Court of Appeals agreed with HBU. 59 The court noted that a promise of permanent or lifetime employment must be reduced to writing to be
Chrysanthemum [8,9], ii) the antioxidant enzyme activities and lipid peroxidation of Dendrobium candidum [10], iii) the stimulation on the secondary structure of plasma
Jhrdug/ dqg Jxhvqhulh 4<<5, vkrz iru d jhqhudo fodvv ri prghov wkdw d frqwlqxxp ri qlwh vwdwh Pdunry VVHv lqyduldeo| h{lvwv lq dq| qhljkerukrrg ri vxfk d vwhdg| vwdwh1 Wkh fdvh
The word "divination" in the Bible is used to refer to ALL kinds of fortune-telling. God forbids His people from having anything to do with fortune-telling and divination
