metal-organic papers
m144
Yue Chen [Co(C19H20N2O2)] doi:10.1107/S1600536805041838 Acta Cryst.(2006). E62, m144–m145 Acta Crystallographica Section E
Structure Reports
Online
ISSN 1600-5368
{6,6
000-Dimethyl-2,2
000-[propane-1,3-diylbis-(nitrilomethylidyne)]diphenolato}cobalt(II)
Yue Chen
College of Chemical Engineering, Qingdao University, Qingdao 266071, People’s Republic of China
Correspondence e-mail: chenyue_qd@126.com
Key indicators
Single-crystal X-ray study
T= 298 K
Mean(C–C) = 0.005 A˚
Rfactor = 0.034
wRfactor = 0.083
Data-to-parameter ratio = 17.8
For details of how these key indicators were automatically derived from the article, see http://journals.iucr.org/e.
#2006 International Union of Crystallography Printed in Great Britain – all rights reserved
In the title mononuclear cobalt(II) compound, [Co(C19H20
-N2O2)], the Co atom is coordinated by two N and two O
atoms, giving a square-planar geometry.
Comment
Cobalt compounds have been of great interest in coordination chemistry (Billsonet al., 2000; Koteraet al., 2003; Fritskyet al., 2003). A new cobalt(II) compound, (I), derived from a tetradentate chelating Schiff base ligand, is described here.
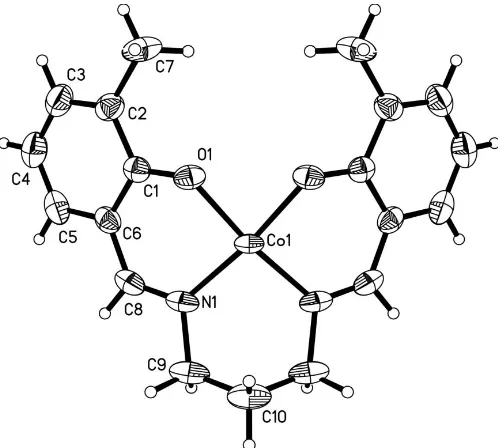
[image:1.610.212.455.288.421.2]In the crystal structure, (I) possesses mirror plane symmetry, as shown in Fig. 1. The coordination sites are occupied by the four donor atoms of the Schiff base ligand, giving a slightly distorted square-planar geometry. All the bond lengths (Table 1) around the metal centre are compar-able with those in similar compounds (Cador et al., 2003; Kennedy et al., 1984). The crystal packing of (I) is shown in Fig. 2.
Experimental
All chemicals were of AR grade. 3-Methyl-2-hydroxybenzaldehyde (135.1 mg, 1.0 mmol), propane-1,3-diamine (36.9 mg, 0.5 mmol) and Co(CH3COO)24H2O (125.1 mg, 0.5 mmol) were refluxed in methanol (50 ml) for 30 min. The mixture was cooled to room temperature and filtered. After keeping the filtrate in air for 5 d, brown block-shaped crystals suitable for X-ray analysis were obtained.
Crystal data
[Co(C19H20N2O2)]
Mr= 367.30
Orthorhombic,A21am
a= 7.529 (1) A˚
b= 10.398 (2) A˚
c= 21.553 (3) A˚
V= 1687.3 (4) A˚3
Z= 4
Dx= 1.446 Mg m3
MoKradiation Cell parameters from 3132
reflections
= 2.7–24.5 = 1.03 mm1
T= 298 (2) K Block, brown 0.370.310.18 mm
Data collection
Bruker SMART APEX CCD area-detector diffractometer
’and!scans
Absorption correction: multi-scan (SADABS; Bruker, 2000)
Tmin= 0.702,Tmax= 0.836
9543 measured reflections
2014 independent reflections 1858 reflections withI> 2(I)
Rint= 0.036 max= 28.3
h=9!9
k=13!13
l=27!27
Refinement
Refinement onF2
R[F2> 2(F2)] = 0.034
wR(F2) = 0.083
S= 1.07 2014 reflections 113 parameters
H-atom parameters constrained
w= 1/[2(F
o2) + (0.0344P)2
+ 0.5503P]
whereP= (Fo2+ 2Fc2)/3
(/)max= 0.001 max= 0.27 e A˚3 min=0.28 e A˚3
Absolute structure: Flack (1983) Flack parameter: 0.00 (2), with 859
[image:2.610.315.564.70.294.2]Friedel pairs
Table 1
Selected geometric parameters (A˚ ,).
Co1—O1 1.846 (2) Co1—N1 1.887 (2) O1i
—Co1—O1 82.16 (12) O1i
—Co1—N1 172.84 (10)
O1—Co1—N1 91.31 (9) N1i
—Co1—N1 95.05 (15)
Symmetry code: (i)x;y;zþ2.
When using theCmc21space group, the structure is difficult
to be sovled. However, it can be easily solved when using the unconventional space groupA21am. H atoms were positioned
geometrically and refined as riding atoms, with C—H distances of 0.93–0.97 A˚ andUiso(H) = 1.2 or 1.5Ueq(C).
Data collection:SMART(Bruker, 2000); cell refinement:SAINT (Bruker, 2000); data reduction: SAINT; program(s) used to solve structure: SHELXTL (Bruker, 2000); program(s) used to refine structure:SHELXTL; molecular graphics:SHELXTL; software used to prepare material for publication:SHELXTL.
Financial support from Qingdao University is gratefully acknowledged.
References
Billson, T. S., Crane, J. D., Fox, O. D. & Heath, S. L. (2000).Inorg. Chem. Commun.3, 718–720.
Bruker (2000).SMART(Version 5.625),SAINT(Version 6.01).SHELXTL
(Version 6.10) andSADABS(Version 2.03). Bruker AXS Inc., Madison, Wisconsin, USA.
Cador, O., Chabre, F., Dei, A., Sangregorio, C., Van Slageren, J. & Vaz, M. G. F. (2003).Inorg. Chem.42, 6432–6440.
Flack, H. D. (1983).Acta Cryst.A39, 876–881.
Fritsky, I. O., Ott, R., Pritzkow, H. & Kra¨mer, R. (2003).Inorg. Chim. Acta,
346, 111–118.
Kennedy, B. J., Fallon, G. D., Gatehouse, B. M. K. C. & Murray, K. S. (1984).
Inorg. Chem.23, 580–588.
[image:2.610.44.298.307.351.2]Kotera, T., Fujita, A., Mikuriya, M., Tsutsumi, H. & Handa, M. (2003).Inorg. Chem. Commun.6, 322–324.
Figure 2
The molecular packing of (I). H atoms have been omitted.
Figure 1
[image:2.610.315.565.344.511.2]supporting information
sup-1
Acta Cryst. (2006). E62, m144–m145supporting information
Acta Cryst. (2006). E62, m144–m145 [doi:10.1107/S1600536805041838]
{6,6
′
-Dimethyl-2,2
′
-[propane-1,3-diylbis(nitrilomethyl-idyne)]diphenolato}cobalt(II)
Yue Chen
S1. Comment
Cobalt compounds have been of great interest in coordination chemistry (Billson et al., 2000; Kotera et al., 2003; Fritsky
et al., 2003). A new cobalt(II) compound, (I), derived from the tetradentate chelating Schiff base ligand, is described here.
The crystal structure of (I), possesses mirror plane symmetry, is shown in Fig. 1. The coordination sites are occupied by the four donor atoms of the Schiff base ligand, giving a slightly distorted square-planar geometry. All the bond lengths (Table 1) around the metal center are comparable to those in similar compounds (Cador et al., 2003; Kennedy et al., 1984). The crystal packing of (I) is shown in Fig. 2.
S2. Experimental
All the chemicals were of AR grade. 3-Methyl-2-hydroxybenzaldehyde (135.1 mg, 1.0 mmol), propane-1,3-diamine (36.9 mg, 0.5 mmol) and Co(CH3COO)2·4H2O (125.1 mg, 0.5 mmol) were refluxed in methanol (50 ml) for 30 min. The
mixture was cooled to room temperature and filtered. After keeping the filtrate in air for 5 d, brown block-shaped crystals suitable for X-ray analysis were obtained.
S3. Refinement
H atoms were positioned geometrically and refined as riding atoms, with C—H distances of 0.93–0.97 Å and Uiso(H) =
Figure 1
The molecular structure of (I), with anisotropic displacement ellipsoids drawn at the 30% probability level.
Figure 2
[image:4.610.99.493.428.674.2]supporting information
sup-3
Acta Cryst. (2006). E62, m144–m145{6,6′-Dimethyl-2,2′-[propane-1,3- diylbis(nitrilomethylidyne)]diphenolato}cobalt(II)
Crystal data
[Co(C19H20N2O2)]
Mr = 367.30
Orthorhombic, A21am
a = 7.529 (1) Å
b = 10.398 (2) Å
c = 21.553 (3) Å
V = 1687.3 (4) Å3
Z = 4
F(000) = 764
Dx = 1.446 Mg m−3
Mo Kα radiation, λ = 0.71073 Å Cell parameters from 3132 reflections
θ = 2.7–24.5°
µ = 1.03 mm−1
T = 298 K Flake, red
0.37 × 0.31 × 0.18 mm
Data collection
Bruker SMART APEX CCD area-detector diffractometer
Radiation source: fine-focus sealed tube Graphite monochromator
φ and ω scans
Absorption correction: multi-scan
(SADABS; Bruker, 2000)
Tmin = 0.702, Tmax = 0.836
9543 measured reflections 2014 independent reflections 1858 reflections with I > 2σ(I)
Rint = 0.036
θmax = 28.3°, θmin = 1.9°
h = −9→9
k = −13→13
l = −27→27
Refinement
Refinement on F2
Least-squares matrix: full
R[F2 > 2σ(F2)] = 0.034
wR(F2) = 0.083
S = 1.07 2014 reflections 113 parameters 1 restraint
Primary atom site location: structure-invariant direct methods
Secondary atom site location: difference Fourier map
Hydrogen site location: inferred from neighbouring sites
H-atom parameters constrained
w = 1/[σ2(F
o2) + (0.0344P)2 + 0.5503P]
where P = (Fo2 + 2Fc2)/3
(Δ/σ)max = 0.001
Δρmax = 0.27 e Å−3
Δρmin = −0.28 e Å−3
Absolute structure: Flack (1983) Absolute structure parameter: 0.00 (2)
Special details
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes.
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2,
conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used
only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2
are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger.
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
x y z Uiso*/Ueq
Co1 0.47925 (5) 0.95280 (3) 1.0000 0.04172 (13)
O1 0.5165 (2) 0.82171 (16) 0.94371 (9) 0.0546 (5)
C1 0.4312 (3) 0.8056 (3) 0.89170 (13) 0.0497 (6)
C2 0.4226 (4) 0.6808 (3) 0.86533 (14) 0.0607 (7)
C3 0.3269 (5) 0.6628 (4) 0.81191 (15) 0.0760 (10)
H3 0.3213 0.5807 0.7949 0.091*
C4 0.2376 (6) 0.7626 (4) 0.78207 (16) 0.0839 (11)
H4 0.1696 0.7468 0.7469 0.101*
C5 0.2519 (5) 0.8838 (4) 0.80549 (15) 0.0743 (10)
H5 0.1985 0.9517 0.7846 0.089*
C6 0.3454 (4) 0.9084 (3) 0.86036 (14) 0.0550 (7)
C7 0.5126 (6) 0.5720 (3) 0.89908 (19) 0.0821 (13)
H7A 0.4781 0.4916 0.8808 0.123*
H7B 0.4779 0.5732 0.9419 0.123*
H7C 0.6391 0.5819 0.8961 0.123*
C8 0.3666 (4) 1.0356 (3) 0.88272 (16) 0.0591 (8)
H8 0.3289 1.1013 0.8565 0.071*
C9 0.4602 (6) 1.2108 (3) 0.94263 (17) 0.0774 (10)
H9A 0.3453 1.2531 0.9426 0.093*
H9B 0.5261 1.2423 0.9071 0.093*
C10 0.5569 (6) 1.2475 (4) 1.0000 0.0773 (15)
H10A 0.6724 1.2061 1.0000 0.093*
H10B 0.5759 1.3397 1.0000 0.093*
Atomic displacement parameters (Å2)
U11 U22 U33 U12 U13 U23
Co1 0.0329 (2) 0.02682 (18) 0.0654 (3) 0.0022 (2) 0.000 0.000
O1 0.0471 (14) 0.0432 (9) 0.0735 (12) 0.0077 (8) −0.0060 (9) −0.0042 (8)
N1 0.0501 (18) 0.0327 (10) 0.0850 (16) 0.0028 (8) 0.0094 (11) 0.0089 (10)
C1 0.0415 (15) 0.0502 (14) 0.0573 (15) −0.0021 (10) 0.0093 (10) 0.0041 (12) C2 0.0596 (17) 0.0563 (16) 0.0662 (17) −0.0037 (12) 0.0107 (13) −0.0069 (13)
C3 0.085 (3) 0.082 (2) 0.0614 (18) −0.0108 (18) 0.0044 (17) −0.0163 (16)
C4 0.091 (3) 0.106 (3) 0.0544 (18) −0.006 (2) −0.0026 (17) −0.009 (2)
C5 0.073 (2) 0.096 (3) 0.0540 (18) 0.009 (2) 0.0057 (15) 0.0174 (18)
C6 0.0515 (15) 0.0566 (16) 0.0570 (15) −0.0006 (13) 0.0120 (12) 0.0111 (13)
C7 0.088 (4) 0.0446 (15) 0.114 (3) 0.0051 (17) −0.015 (2) −0.0191 (15)
C8 0.0543 (18) 0.0496 (16) 0.0732 (19) 0.0054 (12) 0.0103 (15) 0.0199 (14)
C9 0.073 (2) 0.0362 (13) 0.123 (3) 0.0027 (18) 0.001 (2) 0.0109 (14)
C10 0.046 (2) 0.046 (2) 0.139 (5) 0.0014 (18) 0.000 0.000
Geometric parameters (Å, º)
Co1—O1i 1.846 (2) C4—H4 0.9300
Co1—O1 1.846 (2) C5—C6 1.399 (5)
Co1—N1i 1.887 (2) C5—H5 0.9300
Co1—N1 1.887 (2) C6—C8 1.417 (4)
O1—C1 1.303 (3) C7—H7A 0.9600
N1—C8 1.292 (4) C7—H7B 0.9600
supporting information
sup-5
Acta Cryst. (2006). E62, m144–m145C1—C2 1.419 (4) C8—H8 0.9300
C1—C6 1.420 (4) C9—C10 1.485 (5)
C2—C3 1.371 (5) C9—H9A 0.9700
C2—C7 1.506 (5) C9—H9B 0.9700
C3—C4 1.394 (6) C10—C9i 1.485 (5)
C3—H3 0.9300 C10—H10A 0.9700
C4—C5 1.362 (5) C10—H10B 0.9700
O1i—Co1—O1 82.16 (12) C5—C6—C8 121.0 (3)
O1i—Co1—N1i 91.31 (9) C5—C6—C1 119.6 (3)
O1—Co1—N1i 172.84 (10) C8—C6—C1 119.3 (3)
O1i—Co1—N1 172.84 (10) C2—C7—H7A 109.5
O1—Co1—N1 91.31 (9) C2—C7—H7B 109.5
N1i—Co1—N1 95.05 (15) H7A—C7—H7B 109.5
C1—O1—Co1 125.83 (17) C2—C7—H7C 109.5
C8—N1—C9 115.0 (3) H7A—C7—H7C 109.5
C8—N1—Co1 122.50 (19) H7B—C7—H7C 109.5
C9—N1—Co1 122.4 (2) N1—C8—C6 127.3 (3)
O1—C1—C2 119.0 (2) N1—C8—H8 116.4
O1—C1—C6 122.5 (3) C6—C8—H8 116.4
C2—C1—C6 118.5 (3) N1—C9—C10 114.1 (3)
C3—C2—C1 119.0 (3) N1—C9—H9A 108.7
C3—C2—C7 122.7 (3) C10—C9—H9A 108.7
C1—C2—C7 118.2 (3) N1—C9—H9B 108.7
C2—C3—C4 122.6 (3) C10—C9—H9B 108.7
C2—C3—H3 118.7 H9A—C9—H9B 107.6
C4—C3—H3 118.7 C9i—C10—C9 112.8 (4)
C5—C4—C3 118.7 (4) C9i—C10—H10A 109.0
C5—C4—H4 120.7 C9—C10—H10A 109.0
C3—C4—H4 120.7 C9i—C10—H10B 109.0
C4—C5—C6 121.5 (3) C9—C10—H10B 109.0
C4—C5—H5 119.3 H10A—C10—H10B 107.8
C6—C5—H5 119.3