Tandem bispecific neutralizing antibody
eliminates HIV-1 infection in humanized mice
Xilin Wu, … , Paul Zhou, Zhiwei Chen
J Clin Invest.
2018;
128(6)
:2239-2251.
https://doi.org/10.1172/JCI96764
.
Graphical abstract
Research Article
AIDS/HIV
Virology
Find the latest version:
Introduction
Since the discovery of human immunodeficiency virus type 1 (HIV-1) as the causative agent of AIDS in 1983, the search for an effective vaccine or a therapeutic cure has been the top priority in the fight against the expanding HIV/AIDS pandemic. However, because of the tremendous difficulties of HIV-1 vaccine design, generating an appropriate immunogen to elicit broadly neutralizing antibodies (bnAbs) against genetically divergent HIV-1 subtypes (1, 2) has been unsuccessful. With the recent discovery of numerous HIV-1–specific bnAbs (3–9), it has become evident that viral coevo-lution is likely required to drive B cell maturation to induce potent bnAbs during the natural course of infection (2, 10, 11). While there has been an increase in efforts to identify structure-guided nov-el immunogen design for an efficacious vaccine (3, 12–14), using
existing bnAbs as passive immunization is an alternative approach for HIV-1 prophylaxis and immunotherapy (4, 7, 15–20).
Numerous studies have investigated the potency, breadth, crystal structure, and mode of action of selected bnAbs, including their combined use both in vitro and in vivo (16, 21–23). Natural-ly occurring resistant viruses, however, are readiNatural-ly found against these bnAbs when tested individually (9, 21). The bnAb-based monotherapy failed to induce durable suppression of plasma vire-mia as resistant viruses emerged (20, 24). To improve HIV-1 neu-tralization breadth and potency, bispecific bnAbs (bs-bnAbs) have been engineered using the available gene sequences of bnAbs (25–29). In particular, by CrossMAb and knobs-into-holes tech-nologies, bs-bnAb 10E8V2.0/iMab displays exquisite HIV-1–neutral-ization activity in humanized mouse models of HIV-1 prevention and treatment (30). Although bs-bnAbs are promising, their clin-ical development faces large-scale manufacturing challenges and concerns of possible immunogenicity and poor pharmacokinetic properties. Gene transfer of bs-bnAbs may also face several tech-nical challenges. For example, bs-bnAbs generated by the knobs-into-holes method require codelivery of 2 or more genes into the same cell for proportional expression and assembly of antibody light and heavy chains (30). Nevertheless, the recent FDA approv-al of a CD19- and CD3-targeting bispecific antibody for acute B The discovery of an HIV-1 cure remains a medical challenge because the virus rebounds quickly after the cessation of
combination antiretroviral therapy (cART). Here, we investigate the potential of an engineered tandem bispecific broadly neutralizing antibody (bs-bnAb) as an innovative product for HIV-1 prophylactic and therapeutic interventions. We discovered that by preserving 2 single-chain variable fragment (scFv) binding domains of each parental bnAb, a single gene–encoded tandem bs-bnAb, BiIA-SG, displayed substantially improved breadth and potency. BiIA-SG neutralized all 124 HIV-1– pseudotyped viruses tested, including global subtypes/recombinant forms, transmitted/founder viruses, variants not susceptible to parental bnAbs and to many other bnAbs with an average IC50 value of 0.073 μg/ml (range < 0.001–1.03 μg/ml). In humanized mice, an injection of BiIA-SG conferred sterile protection when administered prior to challenges with diverse live HIV-1 stains. Moreover, whereas BiIA-SG delayed viral rebound in a short-term therapeutic setting when combined with cART, a single injection of adeno-associated virus–transferred (AAV-transferred) BiIA-SG gene resulted dose-dependently in prolonged in vivo expression of BiIA-SG, which was associated with complete viremia control and subsequent elimination of infected cells in humanized mice. These results warrant the clinical development of BiIA-SG as a promising bs-bnAb–based biomedical intervention for the prevention and treatment of HIV-1 infection.
Tandem bispecific neutralizing antibody eliminates
HIV-1 infection in humanized mice
Xilin Wu,1,2 Jia Guo,1 Mengyue Niu,1,2 Minghui An,1,3 Li Liu,1,2 Hui Wang,2 Xia Jin,4 Qi Zhang,5 Ka Shing Lam,1 Tongjin Wu,1
Hua Wang,5 Qian Wang,5 Yanhua Du,1 Jingjing Li,1 Lin Cheng,2 Hang Ying Tang,1 Hong Shang,3 Linqi Zhang,5
Paul Zhou,4 and Zhiwei Chen1,2
1AIDS Institute and Department of Microbiology, State Key Laboratory of Emerging Infectious Diseases, Li Ka Shing Faculty of Medicine, The University of Hong Kong, Hong Kong Special Administrative Region, People’s Republic of China. 2The University of Hong Kong AIDS Institute Shenzhen Research Laboratory, Guangdong Key Laboratory of Emerging Infectious Diseases and Shenzhen Key Laboratory of Infection and Immunity, Shenzhen Third People’s Hospital, Shenzhen, Guangdong Province, People’s Republic of China. 3Key Laboratory of AIDS Immunology of National Health and Family Planning Commission, Department of Laboratory Medicine, The First Affiliated Hospital, China Medical University, Shenyang, Liaoning Province, People’s Republic of China. 4Unit of Antiviral Immunity and Genetic Therapy, Institut Pasteur of Shanghai, Chinese Academy of Sciences, Shanghai, People’s Republic of China. 5Comprehensive AIDS Research Center and Collaborative Innovation Center for Diagnosis and Treatment of Infectious Diseases, School of Medicine, Tsinghua University, Beijing, People’s Republic of China.
Related Commentary: p. 2189
Authorship note: XW, JG, MN, MA, and LL contributed equally to this work.
Conflict of interest: A patent on BiIA-SG was awarded to The University of Hong Kong and Shenzhen Third People’s Hospital under ZL201410245945X.
scFv was engineered as a corresponding IA by fusion with human IgG1-Fc to generate IA-PG9, IA-PG16, IA-PGT128, IA-VRC01, and IA-Hu5A8 (Figure 1, A and B). The expression of released soluble IAs was readily detected by Western blot after transient transfec-tion of human 293T cells (Figure 1C). While all IAs exhibited specif-ic anti–HIV-1ADA activity, only IA-PGT128 displayed similar potency and the same sigmoidal slope of 100% neutralization as previously described for the native PGT128 (Figure 1D) (34). Furthermore, we measured the anti–HIV-1 activity of combined IAs in checkerboard experiments (35, 36). We found that IA-PGT128 in combination with IA-Hu5A8 exhibited the best synergistic effect based on com-putational synergy volumes (Figure 1E and Supplemental Figure 1; supplemental material available online with this article; https://doi. org/10.1172/JCI96764DS1). We therefore focused on IA-PGT128 and IA-Hu5A8 for BiIA construction. For mechanism study, we constructed BiIAs by 2 methods. Using the knobs-into-holes meth-od, we generated a double gene–encoded (DG) bispecific IA (BiIA-DG) with the modified IgG1-Fc domain, as previously described (Figure 2A) (25, 37). Using a gene tandem fusion method, we also constructed a single gene–encoded (SG) BiIA (BiIA-SG) through fusion of the PGT128 VL/VH to the N-terminal of IA-Hu5A8 VL/VH in tandem (Figure 2B). As a result, BiIA-SG is structurally unique with 4 scFv binding domains (2 for HIV-1 gp120 and 2 for CD4) as compared with BiIA-DG or other knobs-into-holes bs-bnAbs that contain 2 scFv binding domains (1 for each of the 2 target antigens), as previously described by others (25, 37).
As compared with parental IA-PGT128 and IA-Hu5A8, the expression of BiIA-SG and BiIA-DG was readily detected from transfected 293T cell supernatants with expected sizes deter-mined by Western blot (Figure 2C). To determine their dual spec-ificity, we measured protein binding by 2 methods, ELISA and cell lymphoblastic leukemia has shed light for bs-bnAb–based
immunotherapy (31); allowing this bi-specific antibody to be used for clinical development.
To date, the immunotherapeutic potential of gene-transferred bs-bnAbs has not been investigated in vivo against HIV-1 infection. In this study, we developed a single gene–encoded tandem bispecific immunoadhesin molecule (BiIA), namely BiIA-SG. Engineered immunoadhesin (IA) is an antibody-like molecule, and in this study, IA refers to such molecules that contain the antigen- binding domain of the single-chain variable fragment (scFv) of bnAbs in fusion with the immunoglobulin constant region, includ-ing the hinclud-inge and Fc fragment (e.g., IgG-Fc) but without the con-stant light chain (CL)/concon-stant heavy chain 1 (CH1) (32, 33). We show that BiIA-SG not only displays a potent average IC50 value of 0.073 μg/ml against all 3 panels of 124 genetically divergent HIV-1 strains tested, but also completely prevents diverse live viral challenges in humanized mice. Mechanistically, the improved breadth and potency of the engineered BiIA-SG are associated with the preservation of 2 scFv binding domains of each parental bnAb, which is different from the conventional knobs-into-holes bs-bnAbs. Importantly, gene transfer of BiIA-SG displays the promising activity of eliminating HIV-1–infected cells in many humanized mice. Herein, we provide a proof-of-concept that BiIA-SG is a promising agent for bs-bnAb–based postexposure viremia control and immunotherapy against HIV-1 infection.
Results
Engineering of a single gene–encoded tandem BiIA-SG. Before engi-neering BiIAs, we synthesized codon-optimized scFvs of bnAbs including PG9, PG16, PGT128, VRC01, and Hu5A8 (7–9). The
[image:3.585.42.391.55.342.2]variable light chain (VL)/variable heavy chain (VH) domain of each
and 2 dual-resistant viruses (Figure 3B) as well as 5 VRC01-resis-tant viruses (Supplemental Figure 3) were neutralized by BiIA-SG with improved IC90 or IC50 values. The improved potency of BiIA-SG was also observed with live replicating HIV-1 strains (Figure 3C). To further investigate its antiretroviral potency, we compared BiIA-SG with a panel of 7 bnAbs (VRC01, 3BNC117, PG09, PG16, 10-1074, PGT121, and 10E8) obtained from the NIH AIDS Reagent Program. BiIA-SG consistently exhibited the strongest potency and breadth against 8 genetically divergent and acute HIV-1 pseudoviruses, with average IC50 and IC90 values of 0.023 μg/ml and 0.107 μg/ml, respectively (Figure 4A). BiIA-SG also had similar average IC50 and IC90 values of 0.018 μg/ml and 0.111 μg/ml, respectively, against the NIH global panel of 12 HIV-1 pseudoviruses (Figure 4B) (40). In addition, BiIA-SG has been independently tested in a collaborative laboratory with average IC50 and IC90 values of 0.05 μg/ml and 0.144 μg/ml, respective-surface plasmon resonance (SPR). From the ELISA experiments,
BiIA-SG exhibited slightly reduced binding activity to both sCD4 and gp120 (Figure 2D), whereas BiIA-DG displayed enhanced binding to sCD4 but reduced interaction with gp120. Moreover, by SPR analysis, the KD values indicated that BiIA-SG had a com-parable binding affinity to gp120 but a 229-fold reduced affinity to sCD4 (Table 1 and Supplemental Figure 2). Conversely, BiIA-DG had a comparable binding affinity to sCD4 but a 264-fold reduced affinity to gp120. Our results suggest that 2 PGT128 scFv domains in the single BiIA-SG molecule are necessary for high-affinity binding to gp120.
BiIA-SG displays significantly enhanced potency and breadth against HIV-1. To compare the breadth and potency of BiIA-SG and BiIA-DG with parental IA-PGT128 and IA-Hu5A8, we first tested our University of Hong Kong (HKU) panel of 40 pseudovi-ruses (Supplemental Figure 3), which covered major global HIV-1 subtypes B/B’, CRF01_AE, and C/CRF07_BC/CRF08_BC (34, 38). These 40 pseudoviruses included 8 acute and transmitted/found-er (T/F) strains of various subtypes. VRC01-IgG, one of the sec-ond-generation bnAbs in clinical trials (39), was also included for comparison. Since the molecular weights of IAs are smaller than those of BiIAs and regular antibodies, equimolar concentration (nM) is used for comparison. We found that the mean IC50 and IC90 values of BiIA-SG (1.1 nM/17.9 nM) were significantly bet-ter than those of BiIA-DG (35.1 nM/85.3 nM), IA-PGT128 (69.0 nM/99.6 nM), IA-Hu5A8 (13.0 nM/73.4 nM), and VRC01-IgG (29.2 nM/70.0 nM) (Figure 3A). As a control, the knobs-into-holes BiIA-DG was not significantly better than parental IA-PGT128 and IA-Hu5A8. This finding is consistent with the bs-bnAb PGT128/ iMab generated by the knobs-into-holes and the CrossMAb tech-nologies (30). In contrast, BiIA-SG was able to neutralize 65% (26/40) of pseudoviruses with IC50 values less than 1 nM (~0.15
[image:4.585.45.417.55.326.2]μg/ml). Moreover, 13 IA-PGT128–resistant, 8 IA-Hu5A8–resistant,
Figure 2. Design and characterization of functional BiIAs. (A and B) Sche-matic diagrams show the structures of BiIA-DG and BiIA-SG bearing the scFv binding domain(s) to gp120 (orange VH/VL of PGT128) and to human CD4 (green VH/VL of Hu5A8). (C) The released BiIA-DG, BiIA-SG, and parental IAs were determined in culture super-natants by Western blot analysis under nonreducing conditions (NR, whole left panel) or reducing conditions (R). (D) ELISA binding profiles of BiIA-DG and BiIA-SG to human sCD4 and HIV-1JR-FL gp120, respectively, compared with IA-PGT128, IA-Hu5A8, and VRC01-IgG. Data represent mean ± SD with dupli-cates. All experiments were repeated at least twice.
Table 1. SPR analysis of the binding kinetics/affinity of BiIA-DG, BiIA-SG, and parental IAs to HIV-1JR-FL gp120 and human sCD4 using a Biacore X100
IAs Ka (1/Ms) Kd (1/s) KD (M) Analyte
BiIA-SG 1.208 × 104 1.295 × 10–5 9.197 × 10–10 gp120
BiIA-DG 3.71 × 102 3.718 × 10–5 1.0 × 10–7 gp120
IA-PGT128 1.321 × 104 4.989 × 10–6 3.776 × 10–10 gp120
BiIA-SG 8.26 × 103 5.071 × 10–4 6.139 × 10–8 sCD4
BiIA-DG 4.076 × 103 3.472 × 10–6 8.518 × 10–10 sCD4
IA-Hu5A8 3.607 × 103 9.654 × 10–7 2.676 × 10–10 sCD4
[image:4.585.303.550.616.698.2]IC50 value of 1.32 nM than that of IA-PGT128 (2.36 nM) (Figure 3C, right). Their equimolar combination achieved an IC50 value of 0.79 nM that was still less potent than that of BiIA-SG by 3-fold (0.26 nM). Similar results were confirmed using the NIH global panel of 8 of 12 HIV-1 pseudoviruses based on IC50 and IC90 values (Figure 4B). Overall, BiIA-SG is more potent against both live and genetically divergent viral strains as compared with IA-PGT128, IA-Hu5A8, or their combination in equimolar concentration. Mechanistically, the enhancement of BiIA-SG is probably relat-ed to higher affinity cross-linking interactions with HIV-1 gp120 based on the structural modeling (Supplemental Figure 5) (42).
BiIA-SG confers sterile protection against genetically divergent HIV-1 challenges in humanized mice. To determine the protection efficacy in vivo, we examined the effect of a single injection of BiIA-SG in the HIV-1/NSG-HuPBL model (Figure 5A) (41). A bioreactor product of BiIA-SG generated a peak of 90% purity by size exclusion chromatography analysis (Figure 5B).We then measured the pharmacokinetics and bioavailability of BiIA-SG, which was purified from Chinese hamster ovary (CHO) cells, in 4 healthy NSG-HuPBL mice (2 male [M] and 2 female [F]) and found that its calculated peripheral half-life (t1/2) was approxi-mately 3.1 days (Figure 5C), which fell between the t1/2 values of 5.6 days for Hu5A8 and 2.2 days for PGT128 (7, 16). BiIA-SG ly, against a Tsinghua University panel of 72 HIV-1
pseudovirus-es (Supplemental Figure 4). In this experiment, 21% (15/72) of pseudoviruses had IC50 values less than 0.001 μg/ml. Based on all 3 panels of 124 HIV-1 pseudoviruses tested, our results demon-strate that BiIA-SG is superior to many bnAbs, including BiIA-DG, with a substantially enhanced average IC50 value of 0.073 μg/ml (range < 0.001–1.03 μg/ml) and 100% breadth.
To prepare for in vivo efficacy experiments, the anti–HIV-1 activity of BiIA-SG was evaluated using 2 authentic live viral strains, the R5-tropic HIV-1JR-FL (subtype B) and the R5-tropic T/F virus HIV-1BJZS7 (subtype CRF01_AE). Both HIV-1JR-FL, a tier-2 virus relatively resistant to antibody neutralization (8), and HIV-1BJZS7 established robust systemic and mucosal infections in NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ–human peripheral lymphocyte
[image:5.585.38.391.59.438.2](NSG-HuPBL) mice, as we recently described (41). We found that live HIV-1JR-FL was resistant to IA-Hu5A8 neutralization but was sensitive to IA-PGT128 with an IC50 value of 12.07 nM (Fig-ure 3C, left).The combined equimolar IA-Hu5A8 and IA-PGT128 displayed enhanced neutralizing activity with the IC50 value improved by 6.4-fold to 1.88 nM compared with IA-PGT128. This improved activity, however, was still 6.2-fold less potent than BiIA-SG, which had an IC50 value of 0.30 nM. The neutralization of HIV-1BJZS7 was different because IA-Hu5A8 had a slightly better
most predominant HIV-1 sexually transmitted subtypes (45), we subsequently determined the efficacy of BiIA-SG against live HIV-1BJZS7. Using the same dose and method, we found that BiIA-SG also conferred complete protection against HIV-1BJZS7 (Figure 5, n = 4, 2 M, 2 F). Moreover, all protected animals had undetect-able P24+ T cells in their spleens, lungs, livers, kidneys, stomachs, small intestines, large intestines, and brains by immunofluores-cence staining (Supplemental Figure 6). BiIA-SG could reach the intestinal mucosal sites within 24 hours after a single i.p. injection (Supplemental Figure 7). Our findings demonstrate that soluble BiIA-SG is effective for PrEP against genetically divergent live HIV-1 strains in humanized mice.
[image:6.585.84.514.53.540.2]was then evaluated against HIV-1JR-FL in preexposure prophylaxis (PrEP) experiments using a previously reported dosage (10 mg/ kg, n = 5, 2 M, 3 F) (43, 44). Groups of NSG-HuPBL mice were injected with either BiIA-SG or a placebo (PBS) 1 hour before intraperitoneal (i.p.) inoculation with 10 ng P24 of HIV-1JR-FL (41). All placebo mice (n = 5, 3 M, 2 F) were infected with peak plasma viral loads up to 107 copies/ml (Figure 5D), and exhibit-ed P24 antigenemia (Figure 5E), a trend of CD4+ T cell loss over time (Figure 5F), and P24+ T cells in blood and spleens (Figures 5, G and H) at 2 weeks postinfection (wpi). In contrast, no signs of infection or CD4+ T cell loss were found in 5 of 5 mice that were pretreated with BiIA-SG. Since CRF01_AE is one of the
Single injection of AAV-vectored BiIA-SG eliminates HIV-infected splenocytes in humanized mice. To determine the therapeutic effect of BiIA-SG in vivo, we first compared it with cART in humanized mice. Four days after i.p. challenge with 10 ng P24 of HIV-1, mice were treated with cART (TDF+3TC+RAL regimen, daily), BiIA-SG (400 μg, every 4 days), cART+BiIA-SG combination, or place-bo for 3 weeks (Supplemental Figure 8). When compared with the placebo mice that exhibited high levels of viremia starting from day 4 after viral challenge, all 3 groups of treated mice showed viral suppression with subsequent viral rebound after treatment cessation. Interestingly, the cART+BiIA-SG mice showed a rel-atively delayed viral rebound. These results suggest that short-term treatment (3 weeks) by cART or BiIA-SG or cART+BiIA-SG is insufficient for sustained viral load control or elimination of HIV-1 infection in humanized mice.
We then sought to provide prolonged in vivo BiIA-SG expres-sion using AAV transduction as a monotherapy (15, 17, 46, 47).
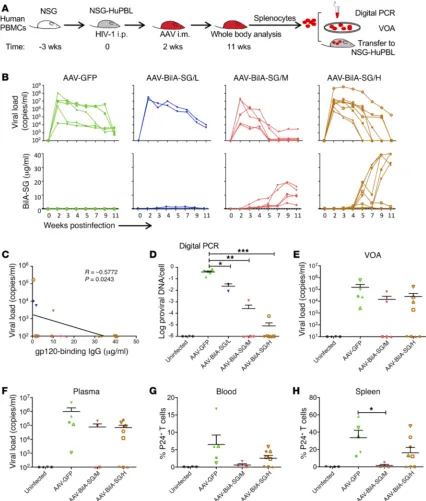
[image:7.585.85.493.54.399.2]BiIA-SG derived from AAV–BiIA-SG–injected mice tends to have an estimated peripheral t1/2 of about 21.7 days (Supplemental Fig-ure 9), which is longer than the t1/2 of 3.1 days of the product puri-fied from CHO cells in vitro (Figure 5C). Two weeks after live HIV-1 infection, groups of NSG-HuPBL mice in 2 separate experiments were treated with a control AAV-GFP (4 × 109 genomic copies [gc] n = 5, 3 M, 2 F) or with a single injection of AAV–BiIA-SG in low-, middle-, or high-dose levels (low dose, 1 × 109 gc, n = 2, 1 M, 1 F; mid-dle dose, 4 × 109 gc, n = 5, 3 M, 2 F; and high dose, 1 × 1011 gc, n = 8, 4 M, 4 F). A group of 4 uninfected mice (2 M, 2 F) served as a nega-tive control (Figure 6A). Infected animals treated with AAV-GFP or low-dose AAV–BiIA-SG exhibited persistent viremia (Figure 6B). Of note, a trend of reduced viral load over time was detected in these mice and it was probably caused by the gradual loss of human CD4+ T cells and corresponding virus-producing cells. In contrast, with increased blood concentrations of BiIA-SG over time, some infected mice treated with middle (4/5) or high doses (5/8) of AAV– Figure 5. Protection of NSG-HuPBL mice against challenges of 2 live and genetically divergent HIV-1 strains. (A) PrEP experimental schedule of BiIA-SG in NSG-HuPBL mice. The dose of BiIA-SG was 10 mg/kg (~200 μg). The HIV-1 inoculum was 10 ng P24. (B) Purity of BiIA-SG by size exclusion chromatogra-phy-high-performance liquid chromatography (SEC-HPLC) analysis. (C) Bioavailability and t1/2 of BiIA-SG in NSG-HuPBL mice. Data represent mean ± SEM. BiIA-SG group, n = 4; PBS group, n = 2. (D) Plasma viral loads among 5 groups of color-coded NSG-HuPBL mice including uninfected control (black, n = 5), HIV-1JR-FL challenged (green, n = 5), BiIA-SG prior to HIV-1JR-FL challenge (red, n = 5), HIV-1BJZS7 challenged (blue, n = 4), and BiIA-SG prior to HIV-1BJZS7 challenge (purple, n = 4). Each line represents data from 1 mouse. (E) Plasma P24 antigenemia by ELISA among the same color-coded groups of animals. The limit of detection was 100 pg/ml. Each line represents data from 1 mouse. (F) The mean percentage of CD4+ T cells in the blood among the 5 groups of animals
tested over time. Data represent mean ± SEM; 2-tailed, unpaired, Student’s t tests were performed. (G and H) The percentage of infected HIV-1 P24+ cells
Figure 6. Therapeutic efficacy of AAV-delivered BiIA-SG in infected NSG-HuPBL mice. (A) Experimental schedule of AAV-delivered BiIA-SG for immuno-therapy of HIV-1JR-FL–challenged humanized mice. A single intramuscular injection of each AAV was performed 2 weeks after HIV-1JR-FL challenge. (B) The top
panel shows the peripheral viral loads tested over time among 4 groups of animals, including AAV-GFP (4 × 109 gc, n = 5), low-dose AAV-BiIA-SG/L (1 × 109
gc, n = 2), middle-dose AAV–BiIA-SG/M (4 × 109 gc, n = 5), and high-dose AAV–BiIA-SG/H (1 × 1011 gc, n = 8). The bottom panel shows peripheral
concen-tration of BiIA-SG expressed over time among the same 4 groups of animals. Each line represents data from 1 mouse. (C) The correlation between plasma viral load and BiIA-SG concentration at 11 wpi among 3 AAV–BiIA-SG groups of animals. Correlation analyses were performed by linear regression using the GraphPad Prism 5.01 program. (D) Proviral loads of peripheral T cells at 11 wpi among uninfected (n = 4) and 4 groups of treated animals by digital PCR. The
y axis represents the amount of DNA copies per cell, plotted as the ratio of HIV-1 DNA to CCR5 copies for each mouse. (E) Before cell adoptive transfer at 11 wpi, donor splenocytes from uninfected and 3 groups of AAV-treated mice were tested by VOA. The y axis indicates viral load copies per milliliter culture supernatants. (F–H) Four weeks after cell adoptive transfer, plasma viral load, the frequency of peripheral P24+ T cells, and the frequency of splenic P24+
al cities in the world (45) but also in small local communities and subpopulations, such as men who have sex with men (48, 49). On the other hand, most bnAbs are not effective against all circulating global HIV-1 strains (Figure 4). For example, PGT128 neutralized 27% and 72% of viruses examined in 2 previous studies (9, 21). In our study, PGT128 was able to neutralize 72.5% of viruses tested. Moreover, 7% of our viruses, including the live HIV-1JR-FL, were not susceptible to Hu5A8 neutralization. In contrast to either PGT128 or Hu5A8 alone, BiIA-SG not only neutralizes 100% of the 3 pan-els of 124 pseudoviruses tested, including the global panel of 12 HIV-1 strains, with substantially improved IC90 values, but also is more potent than many newly discovered bnAbs tested (Figure 4). Critically, our data also demonstrate that BiIA-SG provides com-plete protection against live challenges with genetically divergent HIV-1JR-FL and HIV-1BJZS7. Using the same experimental design and dosage, a previous study showed that bnAb b12 provided 50% pro-tection against HIV-1JR-CSF in humanized mice (44). It is feasible that BiIA-SG can be applied to fight the evolving AIDS epidemic with genetically diverse viruses without the need for prior viral neutral-ization susceptibility tests. It may indeed offer cost effectiveness compared with the combined use of a cocktail of bnAbs (18, 22).
Patients with HIV-1 require lifelong cART mainly because the virus persists in the latent viral reservoir. One of the major tasks in HIV-1 cure research is to discover means to eliminate infected cells, especially those in the reservoir (50). Using HIV-infected humanized mice as a model, a recent study indicated that viral latency is established prior to peak viremia because cART initiat-ed as early as 5 days after infection failinitiat-ed to prevent viral rebound after the cessation of the cART and bnAbs (22). Encouragingly, combination of cART and latency-reversing agents with individual or a cocktail of bnAbs has significantly decreased the latent reser-voir as measured by delayed viral rebound in humanized mice (18, 22, 51, 52). It has been suggested that potent bnAbs might have the advantage to either decrease the half-lives of HIV-1–infected cells in vivo by a FcγR-dependent mechanism or enhance host humoral immunity against HIV-1 probably by forming immune complex for eliciting CD8+ T cell responses (51, 52). Moreover, by conjugating particular dual or ternary bnAbs, the recently developed bispecific 10E8v2.0/iMab and trispecific VRC01/PGDM1400/10E8 achieved exquisite breadth and potency with enhanced protective and ther-apeutic effects (30, 53). In addition, the hinge-modified bs-bnAb 3BNC117/PGT135 showed an improved Fab flexibility and neutral-ization activity (54). In our humanized mouse model, short-term treatment (3 weeks) with either cART or BiIA-SG alone, or with combined cART and BiIA-SG, was insufficient to prevent viral rebound when provided as early as 4 days after live HIV-1 infec-tion. In contrast, a single intramuscular injection of AAV–BiIA-SG at a dose of 4 × 109 gc (middle) or 1 × 1011 gc (high) in 2 separate experiments could produce a sufficient level of functional BiIA-SG in vivo to achieve complete viral load suppression in 3 and 5 ani-mals at 11 wpi, respectively. By 11 wpi, 5 of these 8 controller mice had neither detectable HIV-1 DNA nor any replication-competent viruses in their splenocytes as detected by VOA and cell adoptive transfer experiments. Therefore, in vivo gene transfer of BiIA-SG using an AAV vector was able to eliminate infected splenocytes in 42% (5/12) of the HIV-1–infected humanized mice treated with AAV–BiIA-SG in middle or high doses in the absence of cART or BiIA-SG had viral loads suppressed to undetectable levels from
5–11 wpi (Figure 6B). The level of peripheral BiIA-SG was inverse-ly correlated with the viral load at 11 wpi (Figure 6C). At this end-point, most mice with an undetectable viral load had neither mea-surable proviral DNA as detected by digital PCR (Figure 6D) nor replicating-competent virus by viral outgrowth assays (VOAs) (Fig-ure 6E). Moreover, these animals had undetectable P24+ T cells in their spleens, lungs, intestines, and brains by immunofluorescence staining (Supplemental Figure 10). These results demonstrate that single high-dose injection of AAV–BiIA-SG results in prolonged t1/2 and production of functional BiIA-SG in vivo that in turn eliminates infected cells in humanized mice.
To further validate these results, we conducted cell adoptive transfer experiments using 1 million splenocytes from each treat-ed or AAV-GFP control mouse transferrtreat-ed into individual healthy recipient animals (uninfected, n = 4, 2 M, 2 F; AAV-GFP, n = 5, 3 M, 2 F; AAV–BiIA-SG/M, n = 5, 3 M, 2 F; AAV–BiIA-SG/H, n = 8, 4 M, 4 F). One recipient mouse of the middle-dose group died before analysis. Four weeks after the adoptive transfer, we consistently found that 5 of 12 (42%) recipient mice, which received spleno-cytes from VOA-negative donor mice (2 in the middle- and 3 in the high-dose group), had neither detectable viral load in the serum nor P24+ T cells in the blood or spleens (Figure 6, F–H). In con-trast, live HIV-1 and HIV-infected cells were readily detected in all humanized mice receiving splenocytes from the AAV-GFP group. Our results demonstrate that gene transfer of engineered BiIA-SG leads to the elimination of infected splenocytes, which is likely associated with relatively prolonged in vivo expression of BiIA-SG and VOA negativity in humanized mice.
Discussion
In this study, we report to our knowledge the first single gene– encoded tandem BiIA-SG to facilitate gene transfer of a bs-bnAb– based intervention for HIV-1 prevention and immunotherapy. Each BiIA-SG molecule has 4 scFv-binding domains in total, 2 from each of its parental IAs, PGT128 and Hu5A8. It is structural-ly different from the previousstructural-ly reported bs-bnAb PGT128/iMab that contains only 1 PGT128 and 1 Hu5A8 scFv-binding domain (30). Mechanistically, including 2 PGT128 scFv-binding domains in each BiIA-SG molecule is likely necessary to perform the high- affinity cross-linking interaction with HIV-1 gp120 (42). This notion is in line with the critical topology of PGT128 interaction with the gp120 outer domain at the top surface of the spike (42, 48, 49). In support of this notion, we consistently demonstrated that BiIA-SG is significantly superior to BiIA-DG (Figure 3A), which mimics the PGT128/iMab, for both improved binding to gp120 and enhanced anti–HIV-1 activity. Considering that PGT128 is among the few bnAbs that can achieve a sigmoidal slope of 100% neutralization against HIV-1 infection (34), we believe that the preservation of 2 scFv-binding domains of PGT128 in each BiIA-SG molecule is at least one of the essential mechanisms underly-ing its substantially improved potency and breadth.
individu-pared by automated codon optimization and DNA synthesis accord-ing to published sequences (7–9). Each scFv was constructed as a VL linked to a VH via a 15-mer (Gly4Ser)3 linker (Figure 1). We fused scFvs of these bnAbs to the human immunoglobulin CH2-CH3 (hIgG1-Fc, containing an E333V mutation according to the EU numbering) region. To create secretory antibodies, the secretory signal peptide of tissue plasminogen activator (tPA) was linked to the N-termini of the IAs using PCR-based techniques to enhance the protein expression and release. Two genes encoded BiIA-DG, each with 2 correspond-ing residue mutations to promote the dimerization of electrostatically matched Fc chains using the knobs-into-holes method (25, 37). BiIA-SG was engineered by fusion of scFv-PGT128 to the 5′ end of the scFv-Hu5A8-hIgG1-Fc backbone with a 20-mer (Gly4Ser)4 linker in between (Figure 2). Because we used the original sequence of Hu5A8 but not the ibalizumab (28, 60), we kept the name Hu5A8 in this study.
IA or BiIA expression and purification. 293T cells (ATCC) were
trans-fected with plasmids encoding various IA or BiIA genes and cultured for 72 hours at 37°C in a 5% CO2 incubator after the transfection. IA-con-taining culture supernatants were harvested and centrifuged at 1,000 g for 10 minutes. IAs were purified immediately by affinity chromatogra-phy using Protein G–Agarose (Life Technologies) according to the man-ufacturer’s instructions. The purified IAs and BiIAs were concentrated by an Amicon ultracentrifuge filter device (molecular weight cutoff, 50 kDa; Millipore) to a volume of 0.2 ml in PBS (Life Technologies), and stored at –80°C. Only BiIA-DG was produced by cotransfection of 293T cells using equal amounts of 2 expression vectors.
Western blot analysis. The purity and molecular weights of IAs and
BiIAs were analyzed in 10% SDS-PAGE separating gels with Coomassie Blue G-250 stain (ThermoScientific). The separated proteins were transferred onto PVDF membranes (Millipore) for antibody staining. The reducing and nonreducing conditions refer to the presence and absence of β-mercaptoethanol in the gel-loading buffer. The secondary antibody was an IRDye 800CW-conjugated goat anti–human IgG-Fc domain antibody (catalog 926-32232, Rockland). Protein bands were visualized using the Odyssey Image System (Li-COR).
Binding specificity of IA and BiIA by ELISA. Each well of high-binding
96-well plates (Costar) was coated with 50 ng gp120 or sCD4 overnight. After washing, the plates were blocked for 2 hours with the blocking buffer containing 2% BSA (catalog 10857, Affymetrix), 1 μM EDTA (cat-alog AM9260G, Life Technologies), and 0.05% Tween-PBS (cat(cat-alog Sc-29113, Santa Cruz) and then incubated with serially diluted IA/BiIA for 60 minutes at 37°C. After washing, the HRP-conjugated goat anti– human detection antibody (catalog sc2907, Santa Cruz Biotechnology) was added for 1 hour as previously described (61), followed by washing and addition of 100 μl HRP chromogenic substrate 3,3′,5,5′ -Tetrameth-ylbenzidine (TMB) (catalog 860336, MilliporeSigma). The ODs were measured at 450 nm using the VICTOR3 1420 Multilabel Counter (PerkinElmer). The background values given by incubation of PBS alone in coated wells were subtracted. The positivity was determined when the OD value was 2-fold above the negative controls (e.g., normal serum). All experiments were performed in duplicate.
Surface plasmon resonance (SPR) analysis. The binding kinetics/
affinity of IA and BiIA to gp120 and sCD4 were tested in PBS running buffer at 25°C by SPR analysis using a Biacore X100 machine (GE Healthcare) as previously described (25). HIV-1JR-FL gp120 and sCD4 proteins (30 μg/ml) were covalently coupled to CM5 sensor chips (GE Healthcare) by amine chemistry at pH 5.0, resulting in an
immobiliza-latency-reversing agents. These findings are promising because in combination with cART, a higher dose of 2.5 × 1011 gc AAV
10-1074 and AAV3BNC117 was required to prevent 6 of 7 and 3 of 5 human-ized mice from viral rebound, respectively (22). Moreover, a dose of 1 × 1011 gc AAV
VRC01 was necessary to provide protection in 7 of 10 humanized mice against HIV-1JR-CSF challenge (17). Our data, therefore, demonstrated a proof-of-concept that in the absence of cART or latency inducers, AAV–BiIA-SG monotherapy at a dose of 4 × 109 gc or higher results in HIV-1 control and potentially effec-tively eliminates infected splenocytes in humanized mice.
Besides the cross-linking interactions with HIV-1 gp120 as mentioned above, we cannot exclude other mechanisms that may also contribute to the enhanced anti–HIV activity of BiIA-SG. In regard to neutralizing viruses that are resistant to both PGT128 and Hu5A8, one possible mechanism is that BiIA-SG may confer simultaneous blockade of HIV-1 gp120 and host receptor CD4. Because BiIA-SG has an enhanced anti–HIV potency compared with PGT128 and Hu5A8 in combination, BiIA-SG may also allow the enrichment of the PGT128 domains at the portal of viral entry for more efficient inhibition, as suggested for other bs-bnAbs (28, 30). In addition, it is possible that the binding of BiIA-SG Hu5A8 domains to CD4 increases the local concentration of PGT128 near the cell membrane for enhanced HIV-1 neutralization. This mech-anism is possible because antibody membrane anchoring can increase neutralizing activities of both bnAbs and nonneutralizing anti–HIV antibodies (55, 56). Future structural analysis of BiIA-SG is necessary to answer its mode of action. Comparing the thera-peutic efficacy of the cART+BiIA-SG regimen with the AAV–BiIA-SG regimen, we speculate that AAV–BiIA-AAV–BiIA-SG provides a relatively prolonged and increased in vivo supply of functional BiIA-SG, and therefore, is more effective at the elimination of infected spleno-cytes in humanized mice.
It should be noted that HIV-1–infected humanized NSG-HuP-BL mice do not fully represent infected patients, although this model has been widely used to study bnAb-based HIV-1 immu-notherapy (4, 16, 18). To this end, there are no measurable host immune responses against BiIA-SG and the inoculated viruses. Moreover, besides infected T cells, HIV-1 latency in other cell types remains to be evaluated (41). These caveats need to be care-fully investigated in the future using relevant models. For exam-ple, the humanized bone marrow–liver–thymus (BLT) mouse model could be reconstituted with a broader range of human T cells, B cells, monocytes, and macrophages (57, 58). Human T cell development in transplanted thymus could be observed in the BLT model. Moreover, the humanized myeloid-only mice would be useful to study HIV-1 persistence in tissue-resident macrophages during bnAb immunotherapy (59). Since humanized mice have some limitations, such as incomplete human immune cells, lack of human lymph nodes, and difficulty in generating human antibody and memory T cell immune responses, future BiIA-SG efficacy studies are merited in immune competent nonhuman primate models such as simian human immunodeficiency virus–infected rhesus macaques or directly in human trials (24, 53).
Methods
Engineering of IA and BiIA. Genes encoding single chains (scFv) of
pre-with 5 ml PBS using an 18-gauge needle (catalog NN-1825R, Terumo). Any remaining mucus was then manually extruded into the collection tube. The raw flush was vortexed for 15 seconds and centrifuged for 5 minutes at 16,000 g and the supernatant was separated. The flushed intestines were weighed and homogenized with 100 μl PBS. The homogenate was centrifuged for 5 minutes at 16,000 g and the super-natant was separated. The antibody concentration in small intestine and large intestine mucus and homogenate was detected by ELISA.
Fluorescence-activated cell sorting (FACS) analysis. Blood samples
were collected from the facial veins of mice in Eppendorf tubes con-taining 50 μl anticoagulant (0.5 M EDTA) and were then centrifuged for 5 minutes at 1,150 g in a microcentrifuge. The plasma was stored for future analysis, and the cell pellets were resuspended in 2 ml of 1× RBC lysis buffer (BD Bioscience) and incubated on ice for at least 10 minutes to remove red blood cells. After the lysis, the cells were pel-leted at 1,150 g in a microcentrifuge for 5 minutes at room temperature and stained for 60 minutes at 4°C with 100 μl of a cocktail containing 2 μl anti–human CD3-PB (catalog 300442/UCHT1), 2 μl anti–human CD4-PerCP-Cy5.5 (catalog 317428/OKT4), 2 μl anti–human CD8-PE (catalog 344706/SK1), and 2 μl anti–human CD45 PE/Cy7 antibod-ies (catalog 304016/H130) (all from Biolegend). The samples were washed with PBS supplemented with 2% fetal bovine serum and then were centrifuged at 800 g in a microcentrifuge for 5 minutes. The pel-leted cells were resuspended in 300 μl wash buffer and analyzed on a FACSAria III flow cytometer (Becton-Dickinson). The samples were gated for human CD45 expression before analyzing the T cell subsets based on the CD3, CD4, and CD8 markers within this subset.
Immunofluorescence (IFA) staining of HIV-infected cells in tissues.
Spleen and other tissues were immersed in 10% neutral buffered for-malin (catalog Z2902, MilliporeSigma) for 24 hours. After the forma-lin fixation, the tissues were placed in 70% ethanol (Merck) and sub-sequently embedded with paraffin. Tissue sections (4-μm thick) were used for immunohistochemical staining for HIV-P24 detection using the Kal-1 murine monoclonal antibody (catalog M085701, Dako) as we previously described (41). Confocal images were obtained with a Carl Zeiss LSM 700 microscope using the ZEN 2012 software.
Viral RNA load measurement by quantitative RT-PCR (qRT-PCR).
Viral RNA was extracted using the QIAamp viral RNA mini kit (Qia-gen). Each RNA sample was reverse transcribed to 20 μl cDNA with the RT-PCR Prime Script Kit (Takara). The cDNA (2 μl) was used in a 20 μl qRT-PCR reaction with the TaqMan Universal PCR Master Mix (Life Technologies), a TaqMan probe (5′-FAM−CTCTCT CCTTCT AGCCTC−MGB-3′), and primers designed to target the P17 gene of HIV-1 (5′-TACTGA CGCTCT CGCACC-3′ and 5′-TCTCGA CGCAGG ACTCG-3′). The samples were run in triplicate on an Eppendorf Real-plex4 Mastercycler (Eppendorf). The following cycling conditions were used: 1 cycle of 50°C for 2 minutes, 1 cycle of 95°C for 10 minutes, and 40 cycles of 95°C for 15 seconds and 60°C for 1 minutes. The virus titer was determined by comparison with a standard curve generated using RNA extracted from a serially diluted reference viral stock. The limit of detection was 500 copies per milliliter for HIV-1JR-FL and HIV-1BJZS7.
HIV-1 proviral DNA measurement by digital PCR. Cell-associated
HIV-1 proviral DNA was quantified by QuantStudio 3D digital PCR Sys-tem (Life Technologies). Briefly, total cellular DNA was extracted using the QIAmp DNA Blood Mini Kit (Qiagen) and eluted in 40 μl volume. Purified DNA was quantified for HIV-1 proviral DNA by the QuantStu-dio 3D Digital PCR System using the primers, probe, and reaction condi-tion level of 2,000 response units (RUs) and 4,000 RUs,
respective-ly. BiIA-SG, BiIA-DG, or parental IA was injected through flow cells at 2-fold dilutions starting from 125 nM at a rate of 30 μl/min with 3-min-ute association and 8-min3-min-ute dissociation. The sensor surface was regenerated between each experiment with a 30-second injection of 10 mM glycine•HCl (pH 2.5) at a flow rate of 30 μl/min. Binding curves were plotted after the subtraction of backgrounds. The 2:1 bivalent ana-lyte model was used for the calculations according to the instructions.
HIV-1 neutralization assay. The Global Panel HIV-1 Env clones
were obtained from the NIH AIDS Reagent Program (catalog 12670) (40). The in-house panel of 40 Env clones was generated by us or pro-vided by our collaborator (H Shang, China Medical University, Shen-yang, China). A standard inoculum of 200 TCID50 of each pseudovi-rus was used to test the potency and breadth of various neutralizing antibodies as previously described (62–64). Pseudovirus neutraliza-tion was measured using a luciferase-based assay in GHOST(3)-X4R5 (catalog 3942, NIH AIDS Reagent Program) (35, 38). Each IA and BiIA was tested in duplicate with a 3-fold serial dilution. The 50% and 90% inhibitory concentrations (IC50 and IC90) of each IA and BiIA were calculated to reflect anti–HIV-1 potency. The synergistic effects of paired IAs were measured by a checkerboard synergy assay and calcu-lated using the MacSynergy II software as previously described (36). Accordingly, the synergy volumes of –50 to 50, 50 to 100, and >100 in a synergy plot at the 95% CI were used to define additive, slight synergistic, and highly synergistic effects, respectively. For live HIV-1 neutralization, a standard viral inoculum of 200 TCID50 was used for the assay in TZM-bl cells (catalog 8129, NIH AIDS Reagent Program).
Plasma BiIA-SG quantification by ELISA. For ELISA, 25 ng HIV-1JR-FL
gp120 in coating buffer (pH 9.6) was used to coat 96-well plates (Costar) at 4°C overnight. After washing, the plates were blocked for 1 hour with blocking buffer (5% skim milk in PBS) and then incubated with serially diluted BiIAs or BiIA-containing sera for 60 minutes at 37°C. After wash-ing, goat anti–human secondary antibody conjugated with HRP (catalog sc2907, Santa Cruz Biotechnology) was added for 1 hour, followed by washing and adding 100 μl HRP chromogenic substrate 3,3′,5,5′-TMB (catalog T4444, MilliporeSigma). ODs were measured at 450 nm using the VICTOR3 1420 Multilabel Counter (PerkinElmer). All experiments were performed in duplicate.
Humanized mouse model. Animal procedures that might cause
more than slight pain or distress were performed with appropriate sedation or anesthesia. Immunodeficient NSG mice were purchased from the Jackson Laboratory (catalog 005557). Humanized NSG- HuPBL mice were generated from 4- to 6-week-old NSG mice as we previously described (41).
Purity and half-life of BiIA-SG. The purity of BiIA-SG was
mea-sured by size exclusion–high-performance liquid chromatography (SEC-HPLC). BiIA-SG purified from CHO cells was injected i.p. in to 4 NSG-HuPBL mice at a dose of 20 mg/kg. ELISA was used to measure the serum concentration of BiIA-SG over a period of 7 days. The t1/2 of BiIA-SG was computed as ln (2)/k, where k is a rate constant expressed reciprocally of the x axis time units by the 1-phase decay equation in the GraphPad software.
Intestinal flush and homogenates preparation. A single dose of 10
cells (catalog 240109, Agilent Technologies) were infected with AAV– BiIA-SG. The transgene expression was tested either by FACS on cell surface or by ELISA in cell supernatants after 6 days of infection.
Viral outgrowth assay (VOA). VOA assays were performed as
previ-ously described (66). Briefly, one million splenocytes from AAV-treat-ed mice were seAAV-treat-edAAV-treat-ed into each well of 24-well plates (Corning) in 500
μl RPMI 1640 culture medium (Life Technologies) containing 10% FBS (Life Technologies), and then stimulated by adding 1 μg/ml iono-mycin (catalog I9657, MilliporeSigma) and 0.5 μg/ml phorbol 12-myri-state 13-acetate (PMA, catalog P8139 Sigma-Aldrich). Stimulated splenocytes were cultured for 48 hours at 37°C, 5% CO2, and 100% humidity. Viral RNA copy numbers in culture supernatants were test-ed by qRT-PCR as mentiontest-ed above.
Cell adoptive transfer experiment. One million donor splenocytes
in 100 μl PBS were delivered through i.p. injection into each healthy NSG-HuPBL mouse. Four weeks later, recipient mice were sacri-ficed for analysis.
Statistics. FACS data were analyzed with FlowJo 7.6 software.
Graphs were generated with GraphPad Prism 5.01 software (Graph-Pad Software). Two-tailed Student’s t tests were used for group com-parisons. P < 0.05 was considered statistically significant with neces-sary mean ± SEM or mean ± SD.
Study approval. All animal experimental procedures were
approved by the Committee on the Use of Live Animals in Teaching and Research (CULATR 2786-12) at the Laboratory Animal Unit of The University of Hong Kong. The Laboratory Animal Unit is fully accredited by the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC International).
Author contributions
JG and ZC designed and invented BiIA-SG. XW, JG, MN, and MA performed most experiments and analyzed the data. KSL, YD, JL, TW, LC, HYT, LL, QZ, Hua Wang, and QW provided technical assistance and did animal experiments. Hui Wang, HS, LZ, and PZ provided specimens and viruses. XJ coordinated the bioreactor production. ZC designed and conceived the study, analyzed the data, and wrote the manuscript.
Acknowledgments
We thank the NIH Reagent Program for providing reference bnAbs and the global panel of Env clones. We thank National Natural Science Foundation of China (NSFC) and Hong Kong Research Grants Council (RGC) for the joint grant N_HKU709/11 (to PZ and ZC); Health and Medical Research Fund (HMRF12110952), RGC (HKU5/CRF/13G), Innovation and Technology Fund (ITS/170/17) and the University of Hong Kong Seed Fund (201611160018) (to ZC); China’s National Science and Technology Major Project (2013ZX10001005002001) (to ZC and XJ); NSFC grant 81530065 (to LZ); the Grand Challenge China (81661128042) (to LZ and ZC); the University of Hong Kong Development Fund and The Li Ka Shing Faculty of Medicine Matching Fund (to AIDS Institute); and the San-Ming Project of Medicine in Shenzhen (to ZC and Hui Wang).
Address correspondence to: Zhiwei Chen, AIDS Institute and Department of Microbiology, Laboratory Block L5 40-45, Li Ka Shing Faculty of Medicine, 21 Sassoon Road, Pokfulam, Hong Kong, China. Phone: 852.3917.9825; Email: zchenai@hku.hk.
tions as mentioned above for post-RT HIV-1 RNA quantification. Human CCR5 DNA was quantified with primers (5′-ATGAT TCCTG GGAGA GACGC-3′ and 5′-AGCCA GGACG GTCAC CTT-3′) and the sequence- specific probe (5′-VIC-AACAC AGCCA CCACC CAAGT GATCA- TAMRA-3′). All PCR reactions contained 7.25 μl Quantstudio 3D digi-tal PCR master mix v2 in 14.5 μl reaction volume. HIV-1 proviral load is reported as copies per cell justified by cell numbers from CCR5 copies.
In vivo efficacy of BiIA-SG in humanized mice. For PrEP
experi-ments, 1 day before HIV-1 challenge, blood samples from NSG-HuPBL mice were subjected to flow cytometry to determine the baseline CD4/ CD8 ratios. On the following day, 200 μg (10 mg/kg) BiIA-SG was injected i.p. into NSG-HuPBL mice (44). One hour later the mice were challenged through the i.p. route with 10 ng P24 of live HIV-1JR-FL (466 TCID50) or HIV-1BJZS7 (640 TCID50) diluted in 100 μl PBS (41). These mice were subjected to weekly blood sampling to monitor viral load, CD4/CD8 ratios, and P24+ cells, and finally were sacrificed for the
detection of infected cells throughout the body. For cART treatment experiments, NSG-HuPBL mice were challenged through i.p. route with 10 ng P24 HIV-1BJZS7. Four days after HIV-1 challenge, mice were subjected to 4 different treatments: cART+BiIA-SG, cART, BiIA-SG, and placebo. The cART cocktail was prepared as previously described (22). Individual tablets of tenofovir disoproxil fumarate (TDF, Gilead), Lamivudine (3TC, Shandong Weifang Pharmaceutical Factory Co.), and Raltegravir (RAL, MSD Pharmaceuticals) were triturated into fine powder and suspended in 100 μl PBS. The cART cocktail (TDF 2.46 mg, 3TC 1.48 mg, RAL 1.23 mg) was administered by daily oral gavage to each mouse. BiIA-SG was i.p. injected (400 μg per mouse) every 4 days. All mice were subjected to weekly blood sampling to monitor viral load and were sacrificed 63 days after HIV-1 challenge.
AAV–BiIA-SG production and quantification. AAV–BiIA-SG
1. Kwong PD, Mascola JR, Nabel GJ. Mining the B cell repertoire for broadly neutralizing mono-clonal antibodies to HIV-1. Cell Host Microbe. 2009;6(4):292–294.
2. Haynes BF, et al. HIV-host interactions: impli-cations for vaccine design. Cell Host Microbe. 2016;19(3):292–303.
3. Zhou T, et al. Structural basis for broad and potent neutralization of HIV-1 by antibody VRC01. Science. 2010;329(5993):811–817. 4. Caskey M, et al. Viraemia suppressed in HIV-1-
infected humans by broadly neutralizing anti-body 3BNC117. Nature. 2015;522(7557):487–491. 5. Huang J, et al. Broad and potent neutralization
of HIV-1 by a gp41-specific human antibody. Nature. 2012;491(7424):406–412.
6. Chen YH, Dierich MP. Identification of a second site in HIV-1 gp41 mediating binding to cells. Immunol Lett. 1996;52(2-3):153–156. 7. Reimann KA, et al. A humanized form of a
CD4-specific monoclonal antibody exhibits decreased antigenicity and prolonged plasma half-life in rhesus monkeys while retaining its unique biological and antiviral properties. AIDS Res Hum Retroviruses. 1997;13(11):933–943. 8. Walker LM, et al. Broad and potent
neu-tralizing antibodies from an African donor reveal a new HIV-1 vaccine target. Science. 2009;326(5950):285–289.
9. Walker LM, et al. Broad neutralization coverage of HIV by multiple highly potent antibodies. Nature. 2011;477(7365):466–470.
10. Liao HX, et al. Co-evolution of a broadly neutral-izing HIV-1 antibody and founder virus. Nature. 2013;496(7446):469–476.
11. Gao F, et al. Cooperation of B cell lineages in induction of HIV-1-broadly neutralizing antibod-ies. Cell. 2014;158(3):481–491.
12. Jardine JG, et al. HIV-1 vaccines. Priming a broadly neutralizing antibody response to HIV-1 using a germline-targeting immunogen. Science. 2015;349(6244):156–161.
13. Briney B, et al. Tailored immunogens direct affin-ity maturation toward HIV neutralizing antibod-ies. Cell. 2016;166(6):1459–1470.e11.
14. Diskin R, et al. Increasing the potency and breadth of an HIV antibody by using structure-based rational design. Science. 2011;334(6060):1289–1293.
15. Balazs AB, Chen J, Hong CM, Rao DS, Yang L, Baltimore D. Antibody-based protection against HIV infection by vectored immunoprophylaxis. Nature. 2011;481(7379):81–84.
16. Klein F, et al. HIV therapy by a combination of broadly neutralizing antibodies in humanized mice. Nature. 2012;492(7427):118–122. 17. Balazs AB, et al. Vectored immunoprophylaxis
protects humanized mice from mucosal HIV transmission. Nat Med. 2014;20(3):296–300. 18. Halper-Stromberg A, et al. Broadly neutralizing
antibodies and viral inducers decrease rebound from HIV-1 latent reservoirs in humanized mice. Cell. 2014;158(5):989–999.
19. Pace CS, Fordyce MW, Franco D, Kao CY, Sea-man MS, Ho DD. Anti-CD4 monoclonal antibody ibalizumab exhibits breadth and potency against HIV-1, with natural resistance mediated by the loss of a V5 glycan in envelope. J Acquir Immune
Defic Syndr. 2013;62(1):1–9.
20. Bar KJ, et al. Effect of HIV antibody VRC01 on viral rebound after treatment interruption. N Engl J Med. 2016;375(21):2037–2050.
21. Goo L, Jalalian-Lechak Z, Richardson BA, Over-baugh J. A combination of broadly neutralizing HIV-1 monoclonal antibodies targeting distinct epitopes effectively neutralizes variants found in early infection. J Virol. 2012;86(19):10857–10861. 22. Horwitz JA, et al. HIV-1 suppression and
dura-ble control by combining single broadly neu-tralizing antibodies and antiretroviral drugs in humanized mice. Proc Natl Acad Sci U S A. 2013;110(41):16538–16543.
23. Kong R, et al. Improving neutralization potency and breadth by combining broadly reactive HIV-1 antibodies targeting major neutralization epi-topes. J Virol. 2015;89(5):2659–2671. 24. Scheid JF, et al. HIV-1 antibody 3BNC117
sup-presses viral rebound in humans during treatment interruption. Nature. 2016;535(7613):556–560. 25. Mouquet H, Warncke M, Scheid JF, Seaman MS,
Nussenzweig MC. Enhanced HIV-1 neutraliza-tion by antibody heteroliganeutraliza-tion. Proc Natl Acad Sci U S A. 2012;109(3):875–880.
26. Asokan M, et al. Bispecific antibodies target-ing different epitopes on the HIV-1 envelope exhibit broad and potent neutralization. J Virol. 2015;89(24):12501–12512.
27. Chen W, et al. Exceptionally potent and broadly cross-reactive, bispecific multivalent HIV-1 inhibitors based on single human CD4 and anti-body domains. J Virol. 2014;88(2):1125–1139. 28. Pace CS, et al. Bispecific antibodies directed to CD4 domain 2 and HIV envelope exhibit exceptional breadth and picomolar poten-cy against HIV-1. Proc Natl Acad Sci U S A. 2013;110(33):13540–13545.
29. Sun M, et al. Rational design and characterization of the novel, broad and potent bispecific HIV-1 neutralizing antibody iMabm36. J Acquir Immune Defic Syndr. 2014;66(5):473–483.
30. Huang Y, et al. Engineered bispecific antibodies with exquisite HIV-1-neutralizing activity. Cell. 2016;165(7):1621–1631.
31. Kantarjian H, et al. Blinatumomab versus che-motherapy for advanced acute lymphoblastic leukemia. N Engl J Med. 2017;376(9):836–847. 32. Sekigawa I, Chamow SM, Groopman JE, Byrn RA. CD4 immunoadhesin, but not recombinant soluble CD4, blocks syncytium formation by human immunodeficiency virus type 2-infected lymphoid cells. J Virol. 1990;64(10):5194–5198. 33. Ward RH, et al. Prevention of HIV-1 IIIB infection
in chimpanzees by CD4 immunoadhesin. Nature. 1991;352(6334):434–436.
34. McCoy LE, et al. Incomplete neutralization and deviation from sigmoidal neutralization curves for HIV broadly neutralizing monoclonal anti-bodies. PLoS Pathog. 2015;11(8):e1005110. 35. Kang Y, et al. CCR5 antagonist TD-0680 uses a
novel mechanism for enhanced potency against HIV-1 entry, cell-mediated infection, and a resistant variant. J Biol Chem. 2012;287(20):16499–16509. 36. Prichard MN, Shipman C. A three-dimensional model to analyze drug-drug interactions. Antivi-ral Res. 1990;14(4–5):181–205.
37. Gunasekaran K, et al. Enhancing antibody Fc
heterodimer formation through electrostatic steering effects: applications to bispecific molecules and monovalent IgG. J Biol Chem. 2010;285(25):19637–19646.
38. Todd CA, et al. Development and implementation of an international proficiency testing program for a neutralizing antibody assay for HIV-1 in TZM-bl cells. J Immunol Methods. 2012;375(1–2):57–67. 39. Ledgerwood JE, et al. Safety,
pharmacoki-netics and neutralization of the broadly neu-tralizing HIV-1 human monoclonal antibody VRC01 in healthy adults. Clin Exp Immunol. 2015;182(3):289–301.
40. deCamp A, et al. Global panel of HIV-1 Env ref-erence strains for standardized assessments of vaccine-elicited neutralizing antibodies. J Virol. 2014;88(5):2489–2507.
41. Wu X, et al. Brain invasion by CD4(+) T cells infected with a transmitted/founder HIV-1BJZS7 during acute stage in humanized mice. J Neuro-immune Pharmacol. 2016;11(3):572–583. 42. Pejchal R, et al. A potent and broad neutralizing
antibody recognizes and penetrates the HIV gly-can shield. Science. 2011;334(6059):1097–1103. 43. Mayer KH, et al. Safety, pharmacokinetics, and immunological activities of multiple intravenous or subcutaneous doses of an anti-HIV monoclo-nal antibody, VRC01, administered to HIV- uninfected adults: Results of a phase 1 random-ized trial. PLoS Med. 2017;14(11):e1002435. 44. Gauduin MC, Parren PW, Weir R, Barbas CF,
Burton DR, Koup RA. Passive immunization with a human monoclonal antibody protects hu-PBL-SCID mice against challenge by primary isolates of HIV-1. Nat Med. 1997;3(12):1389–1393. 45. Zhang Y, et al. Dominance of HIV-1 subtype
CRF01_AE in sexually acquired cases leads to a new epidemic in Yunnan province of China. PLoS Med. 2006;3(11):e443.
46. Johnson PR, et al. Vector-mediated gene transfer engenders long-lived neutralizing activity and protection against SIV infection in monkeys. Nat Med. 2009;15(8):901–906.
47. Gardner MR, et al. AAV-expressed eCD4-Ig provides durable protection from multiple SHIV challenges. Nature. 2015;519(7541):87–91. 48. Chen Y, et al. Comprehensive characterization of
the transmitted/founder env genes from a single MSM cohort in China. J Acquir Immune Defic Syndr. 2015;69(4):403–412.
49. Liang J, et al. Community-based HIV-1 early diagnosis and risk behavior analysis of men having sex with men in Hong Kong. PLoS ONE. 2015;10(4):e0125715.
50. Deeks SG, et al. International AIDS Society glob-al scientific strategy: towards an HIV cure 2016. Nat Med. 2016;22(8):839–850.
51. Schoofs T, et al. HIV-1 therapy with monoclonal antibody 3BNC117 elicits host immune responses against HIV-1. Science. 2016;352(6288):997–1001. 52. Lu CL, et al. Enhanced clearance of
HIV-1-infected cells by broadly neutralizing antibodies against HIV-1 in vivo. Science. 2016;352(6288):1001–1004.
Nus-senzweig MC, Ravetch JV. Bispecific anti-HIV-1 antibodies with enhanced breadth and potency. Cell. 2016;165(7):1609–1620.
55. Liu L, et al. The glycosylphosphatidylinositol- anchored variable region of llama heavy chain-only antibody JM4 efficiently blocks both cell-free and T cell-T cell transmission of human immunodeficiency virus type 1. J Virol. 2016;90(23):10642–10659.
56. Liu L, et al. Potent and broad anti-HIV-1 activity exhibited by a glycosyl-phosphatidylinositol- anchored peptide derived from the CDR H3 of broadly neutralizing antibody PG16. J Virol. 2011;85(17):8467–8476.
57. Denton PW, et al. Antiretroviral pre-exposure prophylaxis prevents vaginal transmission of HIV-1 in humanized BLT mice. PLoS Med. 2008;5(1):e16.
58. Sun Z, et al. Intrarectal transmission, systemic infection, and CD4+ T cell depletion in human-ized mice infected with HIV-1. J Exp Med. 2007;204(4):705–714.
59. Honeycutt JB, et al. HIV persistence in tissue macrophages of humanized myeloid-only mice during antiretroviral therapy. Nat Med. 2017;23(5):638–643.
60. Song R, Oren DA, Franco D, Seaman MS, Ho DD. Strategic addition of an N-linked glycan to a mono-clonal antibody improves its HIV-1-neutralizing activity. Nat Biotechnol. 2013;31(11):1047–1052. 61. Klein F, et al. Somatic mutations of the immu-noglobulin framework are generally required for broad and potent HIV-1 neutralization. Cell. 2013;153(1):126–138.
62. Chen Z, Zhou P, Ho DD, Landau NR, Marx PA. Genetically divergent strains of simian
immu-nodeficiency virus use CCR5 as a coreceptor for entry. J Virol. 1997;71(4):2705–2714.
63. Li M, et al. Human immunodeficiency virus type 1 env clones from acute and early subtype B infections for standardized assessments of vaccine-elicited neutralizing antibodies. J Virol. 2005;79(16):10108–10125.
64. Shang H, et al. Genetic and neutralization sen-sitivity of diverse HIV-1 env clones from chron-ically infected patients in China. J Biol Chem. 2011;286(16):14531–14541.
65. Bungiro RD, Sun T, Harrison LM, Shoemaker CB, Cappello M. Mucosal antibody responses in experimental hookworm infection. Parasite Immunol. 2008;30(5):293–303.