VIBRATIONAL SPECTROSCOPIC STUDY AND NBO ANALYSIS ON
ACID
Muthu, S
*1Department of Physics, Sri Venkateswara
2Departments of Physics, Pallavan College of Engineering, Kanchipuram
ARTICLE INFO ABSTRACT
In this work, we report a combined experimental and theoretical study on molecular structure, Vibrational spectra and NBO analysis of
spectra of
frequencies and bonding features of functional method (B3LYP)
from hyper conjugative interactions, charge delocalization has been analyzed using natural bond orbital (NBO) analysis. The calculated HOMO and LUMO energies show that charge transfer occurs within the molecule. The theoretical FT
have been constructed.
INTRODUCTION
Tranexmic acid (TA) is chemically
4-cyclohexane-1-carboxylic acid is a synthetic derivative of the amino acid lysine that exerts its antifibrinolytic effect through the reversible blockade of the lysine binding sites on plasminogen molecules (Thoresen et al., 1981). TA is useful in a wide range of haemorrhagic conditions. The drug reduces postoperative blood losses and transfusion requirements in a number types of surgery (Brown et al., 1997 and Ido 2004), with potential cost and tolerability advantages aprotinin, and appears to reduce rates of mortality and urgent surgery in patients with upper gastrointestinal haemorrhage. TA reduces menstrual blood loss and is a possible alternative to surgery in menorrhagia and has been used successfully to control bleeding in pregnancy. In the present study, FT FT-Raman spectral investigation of TA has been performed using density functional theory (DFT). The redistribution of electron density (ED) in various bonding and antibonding orbital and E(2) energies have been calculated by natural bond orbital (NBO) analysis by DFT method to give clear evidence of stabilization originating from the hyper conjugation of various intra-molecular interactions. The HOMO and LUMO analysis have been used to elucidate information regarding charge transfer within the molecule.
MATERIALS AND METHODS
The compound TA was purchased from sigma
chemical company (USA) with a stated purity of greater than 97% and it was used as such without further purification. The FT-Raman spectrum of TA has been recorded using 1064 nm
*Corresponding author: [email protected]
ISSN:
0975–833X
International Journal of Current Research Vol.
Article History: Received 11th
January, 2011 Received in revised form 18th
February, 2011 Accepted 15th
March, 2011 Published online 14th May 2011
Key Words:
FT-IR , FT-Raman, NBO analysis, HOMO, LUMO and DFT
RESEARCH ARTICLE
VIBRATIONAL SPECTROSCOPIC STUDY AND NBO ANALYSIS ON
TRANEXAMIC
ACID
USING DFT METHOD
Muthu, S1* and Prabhakaran, A2
Department of Physics, Sri Venkateswara College of Engineering, Sriperumbudur-602 105, India Departments of Physics, Pallavan College of Engineering, Kanchipuram-631 502, India
ABSTRACT
In this work, we report a combined experimental and theoretical study on molecular structure, Vibrational spectra and NBO analysis of tranexamic acid (TA).
spectra of TA wear recorded in the solid phase. The molecular geometry, harmonic vibrational frequencies and bonding features of TA in the ground state have been calculated by using density functional method (B3LYP)with standard 6-31G (d,p) basis set. Stability of the
from hyper conjugative interactions, charge delocalization has been analyzed using natural bond orbital (NBO) analysis. The calculated HOMO and LUMO energies show that charge transfer occurs within the molecule. The theoretical FT-IR and FT-Raman spectra for the title molecule have been constructed.
-(aminomethyl) a synthetic derivative of the amino acid lysine that exerts its antifibrinolytic effect through the reversible blockade of the lysine binding sites on 1981). TA is useful in a wide range of haemorrhagic conditions. The drug reduces postoperative blood losses and transfusion requirements in a 1997 and Ido et al.,
2004), with potential cost and tolerability advantages over aprotinin, and appears to reduce rates of mortality and urgent surgery in patients with upper gastrointestinal haemorrhage. TA reduces menstrual blood loss and is a possible alternative to surgery in menorrhagia and has been used successfully to trol bleeding in pregnancy. In the present study, FT-IR, Raman spectral investigation of TA has been performed using density functional theory (DFT). The redistribution of electron density (ED) in various bonding and antibonding s have been calculated by natural bond orbital (NBO) analysis by DFT method to give clear evidence of stabilization originating from the hyper molecular interactions. The HOMO and LUMO analysis have been used to elucidate
ormation regarding charge transfer within the molecule.
was purchased from sigma-Aldrich chemical company (USA) with a stated purity of greater than 97% and it was used as such without further purification. The has been recorded using 1064 nm
line of Nd-YAG laser as excitation wavelength in the region 100-4000 cm-1 on BrukerModel IFS 66 spectrophotometer. The FT-IR spectrum of this compound was recorded in the region 400-4000 cm-1 on IFS 66V Spectrophotometer using KBr pellet technique with a scanning speed of 30 cm and the spectral resolution of 4.0 cm
experimental and calculated FT-IR and FT
shown in Figs. 1 and 2. The spectral measurements were carried out at Sophisticated Analytical Instrumentation Facility (SAIF), IIT, and Chennai.
Computational details
The entire calculations was performed at Density functional theory (DFT) levels with a Pentium Intel (R) core 2 quard 2.40 GHz personal computer using Gaussian 03W (Frisch et al.,2004) program package, invoking gradient geometry optimization (Schlegel, 1982). The optimiz
parameters were used in the vibrational frequencies calculations at the DFT/B3LYP/6
characterize all stationary points as minima. The natural bonding orbitals (NBO) calculation (Glendering
wear performed using NBO 3.1 program as implemented in Gaussian 03W (Frisch et al., 2004) package at DFT/6 31G(d,p) level in order to understand various second interaction between the filled orbitals of one subsystem and vacant of another subsystem, which is measure of intermolecular delocalization or hyper conjugation. The Raman activities (Si) calculated with
converted to relative Raman intensity using Raint program (Michalska, 2003) by the expression:
ternational Journal of Current Research Vol. 33, Issue, 5, pp.078-083, May, 2011
INTERNATIONAL
OF CURRENT RESEARCH
© Copy Right, IJCR, 2011 Academic Journals
TRANEXAMIC
602 105, India 631 502, India
In this work, we report a combined experimental and theoretical study on molecular structure, The FT-Raman and FT-IR geometry, harmonic vibrational in the ground state have been calculated by using density (d,p) basis set. Stability of the molecule arising from hyper conjugative interactions, charge delocalization has been analyzed using natural bond orbital (NBO) analysis. The calculated HOMO and LUMO energies show that charge transfer Raman spectra for the title molecule
YAG laser as excitation wavelength in the region on BrukerModel IFS 66 spectrophotometer. IR spectrum of this compound was recorded in the on IFS 66V Spectrophotometer using KBr pellet technique with a scanning speed of 30 cm-1 min-1 and the spectral resolution of 4.0 cm-1. The observed
IR and FT-Raman spectra are shown in Figs. 1 and 2. The spectral measurements were carried out at Sophisticated Analytical Instrumentation Facility
ulations was performed at Density functional theory (DFT) levels with a Pentium Intel (R) core 2 quard 2.40 GHz personal computer using Gaussian 03W (Frisch et al.,2004) program package, invoking gradient geometry optimization (Schlegel, 1982). The optimized structural parameters were used in the vibrational frequencies calculations at the DFT/B3LYP/6-31G (d,p) level to characterize all stationary points as minima. The natural bonding orbitals (NBO) calculation (Glendering et al., 1998) NBO 3.1 program as implemented in 2004) package at DFT/6-31G(d,p) level in order to understand various second-order interaction between the filled orbitals of one subsystem and vacant of another subsystem, which is measure of the intermolecular delocalization or hyper conjugation. The Raman activities (Si) calculated with Gaussian 03were converted to relative Raman intensity using Raint program
INTERNATIONAL JOURNAL OF CURRENT RESEARCH
Fig.1. The atom numbering for TA molecule
I
i=
10
-12
-
i
1
/
i
S
iWhere Ii is the relative Raman intensity, Si, the Raman activities, i is the wave number of normal modes and
denotes the wave number of the excitation laser(Michalska, 2005).
RESULTS AND DISCUSSION
Molecular geometry
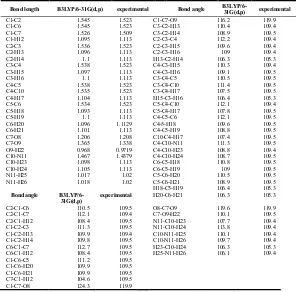
The labeling of atoms in TA is given in Fig 3. The optimized geometrical parameters (bond length and bond angles) by DFT/B3LYP with 6-31G (d,p) basis set are listed in Table 1, a general priority for reproducing the experimental bond length
Fig. 3. FT-Raman spectra of TA calculated and experimental
taken from the Ref (Carl kemnitz, 2002) is not present among DFT/B3LYP levels. However, all the bond and bond angles computed with the DFT/B3LYP level shows excellent agreement with available experimental results.
Vibrational assignments
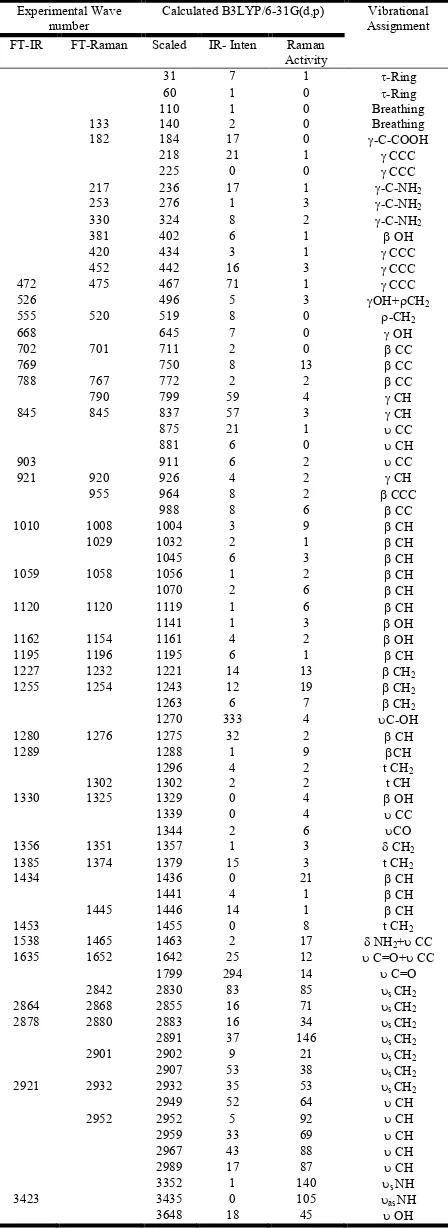
Vibrational spectral assignments have been performed on the recorded FT-IR and FT-Raman spectra based on the theoretically predicted wave numbers by density functional method (B3LYP) using 6-31G (d,p) basis set and have been collected in Table 2. Comparison of the frequencies calculated at B3LYP method with experimental values reveals the overestimation of the calculated vibrational modes due to neglect of anharmonicity in real system. In our study we have followed scaling factor for B3LYP/6-31G (d,p); 0.9608 (Scoott et al., 1996).
C-H Vibration
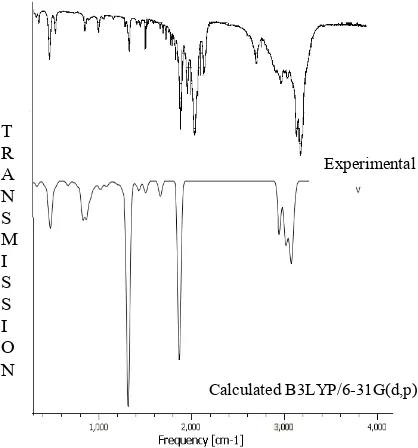
The hetero aromatic structure shows the presence of C-H stretching vibration in the region 3100-3000 cm-1 which is the
characteristic region for the ready identification of C-H stretching vibration (Varsanyi, 1973). In this region, the bands are not affected appreciably by the nature of substituents. The vibration assigned by chemcraft program package at 2971 to 2952 cm-1 by B3LYP/6-31G (d,p) levels, show good agreement with weak FT-Raman band at 2952 cm-1 wear assigned to the C-H stretching vibration in the aromatic ring. In general in-plane and out-of-plane aromatic C-H deformations occur in the region 1300-1000 cm-1 and 600-1000 cm-1 respectively. The C-H in-plane bending vibration computed at 1057, 1196 and 1277 cm-1 by B3LYP/6-31G(d,p) method shows good agreement with experimental values. The C-H out of plane bending vibration band appears in FT-IR at 845 cm-1 and in FT-Raman at 852 cm-1 shows good agreement with computed DFT (B3LYP) method at 838 cm-1. In general C-H vibration computed by both methods shows good agreement with experimental observation as well as literature data.
T R A N S M I S S I O N
Experimental
Calculated B3LYP/6-31G(d,p)
Fig. 2. FTIR spectra of TA calculated and experimental
R
a
m
a
n
[image:2.612.75.284.283.507.2]Fig. 4. The atomic orbital HOMO – LUMO composition of the frontier molecular orbital for TA
O-H vibrations
The O-H group gives rise to three vibration (stretching, in-plane bending and out-of-in-plane bending vibration).The O-H group vibrations are likely to be most sensitive to the
environment, so they show pronounced shifts in the spectra of the hydrogen bonded species. The hydroxyl stretching vibrations are generally (Sajan et al., 2006) observed in the region around 3500 cm-1. In the case of the un-substituted phenols it has been shown that the frequency of O-H stretching vibration in the gas phase is 3657 cm-1 (Michalska
et al., 1996). Similarly in our case a strong FT-IR band at 3423 cm-1 is assigned to O-H stretching vibration. The hydrogen bonding effect through hydroxyl group leads to dimmer conformation OH stretching mode calculated at 3652 cm-1 which is much closer to the FT-IR experimental observation at 3423 cm-1.
The O-H in-plane bending vibration in the phenols, in general lies in the region 1150-1250 cm-1 and is not much affected due to hydrogen bonding unlike to stretching and out-of-plane bending frequencies. The medium band in FT-IR spectrum at 1162 and 1195 cm-1 is assigned O-H in-plane bending vibration. Theoretically computed value at 1143 and 1163 cm-1 by B3LYP method shows good agreement with recorded spectrum. The O-H out-of-plane bending mode for the free molecule lies below 300 cm-1 and it is beyond the infrared spectral range of the present investigation. However, for the associated molecule (Varsanyi, 1973) the O-H out-of plane bending mode lies in the region 517-710 cm-1 in both intermolecular and intramolecular association, the frequency is at a high value than in free O-H. In our present investigation a strong band observed in FT-IR spectrum at 555 cm-1 is
assigned to O-H out-of-plan bending vibration, the theoretically computed value by B3LYP shows the same kind of vibration at 520 and 646 cm-1 are assigned to O-H out-of-plane bending vibration.
LUMO PLOT First excited state
E LUMO =0.369 ev
∆E = -11.692
E HOMO =-1.323ev
HOMO PLOT Ground state
Table 1. Comparison between the calculated DFT and experimental values of geometrical parameters for TA
Bond length B3LYP/6-31G(d,p) experimental Bond angle
B3LYP/6-31G(d,p) experimental
C1-C2 1.545 1.523 C1-C7-O9 116.2 119.9 C1-C6 1.545 1.523 C3-C2-H13 110.4 109.4 C1-C7 1.526 1.509 C3-C2-H14 108.9 109.5 C1-H12 1.095 1.113 C2-C3-C4 112.2 109.4 C2-C3 1.536 1.523 C2-C3-H15 109.6 109.4 C2-H13 1.096 1.113 C2-C3-H16 109 109.4 C2-H14 1.1 1.113 H13-C2-H14 106.3 105.3 C3-C4 1.538 1.523 C4-C3-H15 110.3 109.4 C3-H15 1.097 1.113 C4-C3-H16 109.1 109.5 C3-H16 1.1 1.113 C3-C4-C5 110.5 109.5 C4-C5 1.538 1.523 C3-C4-C10 111.4 109.5 C4-C10 1.535 1.523 C3-C4-H17 107.5 109.5 C4-H17 1.104 1.113 H15-C3-H16 106.4 105.3 C5-C6 1.534 1.523 C5-C4-C10 112.1 109.4 C5-H18 1.093 1.113 C5-C4-H17 107.8 109.5 C5-H19 1.1 1.113 C4-C5-C6 112.1 109.5 C6-H20 1.096 1.1129 C4-5-H18 109.6 109.5 C6-H21 1.101 1.113 C4-C5-H19 108.8 109.5 C7-O8 1.206 1.208 C10-C4-H17 107.4 109.5 C7-O9 1.365 1.338 C4-C10-N11 111.3 109.5 O9-H22 0.968 0.9719 C4-C10-H23 108.8 109.4 C10-N11 1.467 1.4379 C4-C10-H24 108.7 109.5 C10-H23 1.098 1.113 C6-C5-H18 110.8 109.5 C10-H24 1.105 1.113 C6-C5-H19 109 109.5 N11-H25 1.017 1.02 C5-C6-H20 110.5 109.5 N11-H26 1.018 1.02 C5-C6-H21 108.9 109.5 H18-C5-H19 106.4 105.3
Bond angle
B3LYP/6-31G(d,p)
experimental H20-C6-H21 106.3 105.3
C2-C1-C6 110.5 109.5 O8-C7-O9 119.6 119.9 C2-C1-C7 112.1 109.4 C7-O9-H22 110.1 109.5 C2-C1-H12 108.4 109.5 N11-C10-H23 107.7 109.4 C1-C2-C3 111.3 109.5 N11-C10-H24 113.8 109.4 C1-C2-H13 109.9 109.4 C10-N11-H25 110.1 109.4 C1-C2-H14 109.8 109.5 C10-N11-H26 109.7 109.4 C6-C1-C7 112.7 109.5 H23-C10-H24 106.3 105.3 C6-C1-H12 108.4 109.5 H25-N11-H26 106.1 109.4 C1-C6-C5 111.2 109.5
[image:3.612.153.449.394.686.2]Table 3. Second order perturbation theory analysis of Fork matrix in NBO basis for TA
Table 4. HOMO LUMO energy calculated by B3LYP/6-31G(d,p) method
Method B3LYP/6-31G(d,p) HOMO
LUMO Energy gap (∆E)
-11.323eV 0.369eV -11.692eV
C-C ring stretching
In benzonitriles, the distance between two carbon atoms changes the ring angles because of its substituents groups such .as cyanogens. There are six equivalent C-C bonds in benzene and consequently there will be six C-C stretching vibrations. The bands are observed at 1538 and 1632 cm-1 in FT-IR are identified as C-C stretching vibrations. The same vibration appear in the FT-Raman spectrum at 1652 cm-1. The theoretically scaled C-C stretching vibrations by DFT method are at 1642-1 shows excellent agreement with recorded FT-IR and FT-Raman spectral data. The ring in-plane vibrations have given rise to weak bands across the low frequency region, that is to say, below 1000cm-1 the bands at 751 and 989 cm-1 have been assigned to C-C in-plane bending vibrations.
CH2 bending vibrations
In the present study, the band around 1280cm-1 in FT-IR is assigned to CH2 wagging mode which agrees with the result
(1281cm-1 ) of (Matulkova et al) in the case of 4-aminotriazole adipic acid (4atadip). The harmonic frequencies calculated by B3LYP/6-31G(d,p) method falling in the region 1340-1277 cm-1 , which are in agreement with the earlier report (Matulkova et al). The calculated frequencies 1297 and 1359 cm-1 are assigned to twisting of CH2 . Matulkova et al.
reported the CH2 twisting frequency observed in the range
1223-1314 cm-1 of 4atadip. In the present work CH2 twisting
vibration is assigned to medium Raman band at 1276 and 1302 cm-1. However , the DFT value is in agreement with the experimental value. The band appeared around 1356 cm-1 in FT-IR is assigned to CH2 scissoring mode. The computed CH2
rocking mode is appears at 520 cm-1 in DFT. The FT-IR band at 526 cm-1 and Raman band at 535 cm-1 are assigned to CH2
rocking mode, which agree favourably with Matulkova et at. and also find support from theoretical value. Billes et al. assigned CH-in-plane bending vibration appeared at 1419, 1114, 1411 and 1126 cm-1, respectively in 1H-1,2,3-and 1D-1,2,3-triazoles. In view of above bands appeared at 1435 cm-1
Donor(i) Type ED/e Acceptor(j) Type ED/e E(2) (kcal/mol)
C4-C10 σ 1.96 C2-C3 σ* .015 2.16
C3-C4 σ* .019 0.99
C4-C5 σ* 0.19 1.07
C4-H17 σ* 0.02 0.55
N11-H25 σ* 0.006 2.13
N11-H25 σ 1.98 C4-C10 σ* 0.02 3.09
N11-H26 σ 1.98 C10-H23 σ* 0.018 3.03
LP(1)N11 1.96 C4-C10 σ* 0.02 0.87
C10-H23 σ* 0.01 1.12
C10-H24 σ* 0.03 7.39
LP(1)O9 1.82 C1-C7 σ* 0.06 7.46
C7-O8 σ* 0.01 2.24
LP(2)O9 1.82 C7-C8 σ* 0.20 47.40
LP(1)O8 1.97 C1-C7 σ* 0.06 2.61
C7-O9 σ* 0.09 1.01
LP(2)O8 1.855 C1-C7 σ* 0.022 17.07
[image:4.612.60.284.78.692.2]C7-O9 σ* 0.09 34.57
Table 2. Vibrational wavenumbers obtained for TA at B3LYP/6-31G(d,p) and compared with experimental value
Experimental Wave number
Calculated B3LYP/6-31G(d,p) Vibrational Assignment FT-IR FT-Raman Scaled IR- Inten Raman
Activity
31 7 1 -Ring
60 1 0 -Ring
110 1 0 Breathing
133 140 2 0 Breathing
182 184 17 0 -C-COOH
218 21 1 CCC
225 0 0 CCC
217 236 17 1 -C-NH2
253 276 1 3 -C-NH2
330 324 8 2 -C-NH2
381 402 6 1 OH
420 434 3 1 CCC
452 442 16 3 CCC
472 475 467 71 1 CCC
526 496 5 3 OH+CH2
555 520 519 8 0 -CH2
668 645 7 0 OH
702 701 711 2 0 CC
769 750 8 13 CC
788 767 772 2 2 CC
790 799 59 4 CH
845 845 837 57 3 CH
875 21 1 CC
881 6 0 CH
903 911 6 2 CC
921 920 926 4 2 CH
955 964 8 2 CCC
988 8 6 CC
1010 1008 1004 3 9 CH
1029 1032 2 1 CH
1045 6 3 CH
1059 1058 1056 1 2 CH
1070 2 6 CH
1120 1120 1119 1 6 CH
1141 1 3 OH
1162 1154 1161 4 2 OH
1195 1196 1195 6 1 CH
1227 1232 1221 14 13 CH2
1255 1254 1243 12 19 CH2
1263 6 7 CH2
1270 333 4 C-OH
1280 1276 1275 32 2 CH
1289 1288 1 9 CH
1296 4 2 t CH2
1302 1302 2 2 t CH
1330 1325 1329 0 4 OH
1339 0 4 CC
1344 2 6 CO
1356 1351 1357 1 3 CH2
1385 1374 1379 15 3 t CH2
1434 1436 0 21 CH
1441 4 1 CH
1445 1446 14 1 CH
1453 1455 0 8 t CH2
1538 1465 1463 2 17 NH2+ CC
1635 1652 1642 25 12 C=O+ CC
1799 294 14 C=O
2842 2830 83 85 s CH2
2864 2868 2855 16 71 s CH2
2878 2880 2883 16 34 s CH2
2891 37 146 s CH2
2901 2902 9 21 s CH2
2907 53 38 s CH2
2921 2932 2932 35 53 s CH2
2949 52 64 CH
2952 2952 5 92 CH
2959 33 69 CH
2967 43 88 CH
2989 17 87 CH
3352 1 140 s NH
3423 3435 0 105 as NH
3648 18 45 OH
- stretching; s- symmetric stretching; as- asymmetric stretching; - in-plane bending; - out of plane bending;
[image:4.612.361.505.258.298.2]in FT-Raman and 1445 in FT-IR spectrum are assigned to C-H in plane bending mode. The calculated frequencies in the region 828-956 cm-1 for C-H out-of- plane bending fall in the FT-IR and FT-Raman values of 845-920 cm-1.
NH2 vibration
The molecule under investigation possesses only one NH2
group and hence one expects one symmetric and one asymmetric N-H stretching vibration in NH2 group. In all the
primary aromatic amines, the N-H stretching frequency occurs in the region 3300-3500 cm-1 (Bellamy, 1980). Hence the weak bands in FT-IR spectrum wear located at 3320-3232 cm
-1
assingned to N-H asymmetric and symmetric stretching vibration, respectively in NH2 group. These assignments agree
well with the earlier report (Bellamy, 1980). The scaled NH2
asymmetric and symmetric stretching are in the range 3439-3356 cm-1 in B3LYP/6-31G(d,p). The computed NH2
scissoring vibration at 1465 cm-1 in B3LYP/6-31G(d,p) is in agreement with the expected experimental value at 1465 cm-1. The C-NH2 out-of-plane and in-plane bending vibration at 253
and 381 cm-1 observed at FT-Raman spectrum agree well with theoretically obtained values using B3LYP/6-31G (d,p).
COOH vibration
Carboxylic acid dimmer is formed by strong hydrogen bonding in the solid and liquid state vibrational analysis of carboxylic acid group is made on the basis of carbonyl group and hydroxyl group. The C=O stretch of carboxylic acid is identical to the C=O stretch in ketones, which is expected in the region 1740-1660 cm-1 (Vein et al., 1991). The C=O bond formed by Pπ- Pπ between C and O intermolecular hydrogen
bonding reduces the frequencies of the C=O stretching absorption to a greater degree than does intermolecular H bonding because of the different electro-negativities of C and O, the bonding are not equally distributed between the two atoms. The loan pair of electrons on oxygen also determines the nature of the carbonyl groups. In our present study a strong band observed in FT-IR spectrum 1635 cm-1 is assigned to C=O stretching vibrations, which show good agreement with B3LYP scaled value at 1642 cm-1. Two other characteristic carboxylic group vibrations are C-O stretching and C-O bending vibrations. They are expected in the region 1140-395 cm-1 and 1700-875 cm-1 depending on whether monomeric, dimeric or other hydrogen bonded species are present. Generally the C-O stretching mode appears at lower frequencies than C-O bending vibration.
NBO Analysis
Natural bond orbital analysis gives the most accurate possible natural Lewis structure picture of Ø because all orbital are mathematically chosen to include the highest possible percentage of the electron density. Interaction between both filled and virtual orbital space information is correctly explain by the NBO analysis, it could enhance the analysis of intra and inter molecular interaction. The second order Fock matrix was carried out to evaluate donor (i)-acceptor (j) i.e. donar level bonds to acceptor level bond interaction in the NBO analysis (Szafran et al., 2007). The result of interaction is a loss of occupancy from the concentration of electron NBO of the idealized Lewis structure into an empty non-Lewis orbital. For
each donor (i) and acceptor (j), the stabilization energy E(2) associates with the delocalization i→ j is estimated as
E(2) =ΔEij=q
i(F(i,j)2/( εj – εi)
Where qi is the donor orbital occupancy, εi - and εj are
diagonal element and F(i,j) is the off diagonal NBO Fock matrix element. Natural bond orbital analyses provide an efficient method for interaction among bond, and also provide a convenient basis for investigation charge transfer or conjugative interaction in molecular systems. Some electron donor orbital, acceptor orbital and the interaction stabilization energy resulted from the second order micro-disturbance theory are reported (James et al., 2006 and Jun-na et al.,
2005). The larger E(2) value the more intensive is the interaction between electron donors and acceptor i.e. the more donation tendency from electron donors to electron acceptors and the greater the extent of conjugation of the whole system (Sebastian et al., 2010). Delocalization of electron density between occupied Lewis-type (bond or lone pair) NBO orbitals and formally unoccupied (anti bond or rydberg) non Lewis NBO orbital correspond to astabilizing donor-acceptor interaction. NBO analysis has been performed on the Ta molecule at the DFT/B3LYP/6-31G(d,p) level in order to elucidate, the intra molecular rehybridization and delocalization of electron density within the molecule. The LP(2) O9 is seen to be lowest-occupancy and to be primarily delocalized into ant bond C7-O8. The E(2) values and types of
transition as shown Table 3.
HOMO and LUMO analysis
Highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) and very important parameters for quantum chemistry. We can determine the way the molecule interacts with other species. Hence, they are called the frontier orbitals. HOMO, which can be thought the outermost orbital containing electrons, trends to give these electrons such as an electron donor. On the other hand; LUMO can be thought the innermost orbital containing free places to accept electrons (Gece et al., 2008).The HOMO and LUMO energy calculated by B3LYP /6-31G(d,p) method are shown in Table 4. This electronic transition absorption corresponds to the transition from the ground to the first excited state and is mainly described by an electron excitation from the HOMO to the LUMO. The HOMO is located over the NH2 group, the
HOMO > LUMO transition implies an electron density transfer to ring and acid group. The atomic compositions of the frontier molecular orbital are shown in Fig. 4. The calculated self-consistent field (SCF) energy of TA is -519.11 A.U
Conclusion
coincide with experimentally observed FT-IR, FT-Raman. NBO reflects the charge transfer within the molecule. HOMO and LUMO orbitals have been visualized.
REFERENCE
Bellamy, L.J. 1980. The infrared spectra of complx molecule vol 2
Billes, F, Endredi, H, and Keresztury, G, 2000. Vibrational spectroscopy of triazoles and tetrazole. J. Mol. Struct.,
(Theochem). 530 : 183.
Brown, R.S, Thwaites, B.K, Mongan, P.D, 1997. Tranexamic acid is effective in decreasing postoperative bleeding and transfusions in primary coronary artery bypass operations: a double-blind, randomized, placebo-controlled trial.
Anesth Analg, J Obstet Gynecol., 85: 963-70. Carl Kemnitz, 2002. Chemoffice ultra 10, Trial version.
Frisch, M. J, et al., 2004. Gaussian 03W, Revision C.02.
Wallingford: Gaussian Inc.,
Gece, G, 2008. The use of quantum chemical methods in corrosion inhibitor studies. Corros. Sci., 50: 2981.
Glendering, E.D, Read, A.E, Carpenter, J.E, and Weinhold, F, 1998. NBO Version 3.1. TCI, University of Wisconcin, Madison..
Ido, K, Neo, M, Asada, Y et al., 2000. Reduction of blood loss using tranexamic acid in total knee and hip arthroplasties.
Arch Orthop Trauma Surg., 120:518-20
James, C, Amal Raj, A, Rehunathan, R, Hubert Joe, I, Jayakumar. V.S. 2006. J. Raman Spectrosc, 379 1381 Jun-na, L, Zhi-rang, C, Shen-fang, Y and Zhejiag, J, 2005.
Univ. Sci. 6B (2005) 584
Matulkova, I, Nemec, I, Teubner, K, Nemec, P, and Micka, Z, 2008. Novel compound of 4-amino-1,2,4-triazole with dicarboxylic acids - crystal structures, vibrational spectra and non-linear optical properties. J. Mol. Struct., 873 : 46. Michalska, D and Wysokinski, R, 2005. Chem. The prediction
of Raman spectra of platinum(II) anticancer drugs by density functional theory, Phys. Lett, 403: 211.
Michalska, D, 2003. Raint Program, Wroclaw University of Technology.
Michalska,D, Bienko, D.C,. Bienko, A.J.A , and Latajka, Z, 2008. Density Functional, Hartree-Fock and MP2 studies on the vibrational spectrum of phenol. J. Phys Chem., 100: 1186.
Sajan,D, Hubert Joe,I, Jayakumar,V.S, and Zaleshki,J, 2006. Structural and electronic contributions to hyperpolarizability in methyl p-hydroxy benzoate. J. Mol. Struct., 785: 43
Schlegel, H. B, 1982. Optimization of equilibrium geometries and transition structures. J. Comput. Chem., 3: 214-218. Scott, A.P, Radom, L, 1996. Harmonic vibrational
frequencies: an evaluation of Hartree-Fock, Møller-Plesset, quadratic configuration interaction, density functional theory, and semiempirical scale factors. J. Phys. Chem.,
100: 16502
Sebastian, S, and Sundaragnesan, N, 2010. The spectroscopic (FT-IR, FT-IR gas phase, FT-Raman and UV) and NBO analysis of 4-Hydroxypiperidine by density functional method. Spectrochim Acta, A 75 : 941.
Szafran, M, Komasa, A,and Adamska, E.B, 2007. Crystal and molecular structure of 4-carboxypiperidinium chloride (4-piperidinecarboxylic acid hydrochloride J. Mol. Struct. (THEOCHEM) 827: 101.
Thorsen S, Clemmenson I, Sottrup-Jensen L et al.1981. Adsorption to fibrin of native fragments of known primary structure from human plasminogen. Biochim Biohys Acta.,
668: 377-87.
Varsanyi , G, 1973. Assignment of vibrational spectra of seven hundred Benzene derivatives, 1/2, Academic kiaclo, Budapest.
Vein, D.L, Colthup, N.B, Fateley, W.G and Grasselli, J.G,1991. The Handbook of Infrared and Raman Characteristic Frequencies of Organic Molecules, Academic press, San Diego.