High Performance Computing with Coarse Grained Model of
Biological Macromolecules
Emilia Lubecka1,2, Adam Sieradzan2, Cezary Czaplewski2, Pawe l Krupa3,
Adam Liwo2
c
The Authors 2018. This paper is published with open access at SuperFri.org
The Unified Coarse Grained Model of biological macromolecules (UCGM) that is being de-veloped in our laboratory is a model designed to carry out large-scale simulations of biological macromolecules. The simplified chain representation used in the model allows to obtain 3-4 orders of magnitude extention of the time-scale of simulations, compared to that of all-atom simulations. Unlike most of the other coarse-grained force fields, UCGM is a physics-based force field, indepen-dent of structural databases and applicable to treat non-standard systems. In this communication, the efficiency and scalability of the new version of UCGM package with Fortran 90, with two par-allelization levels: coarse-grained and fine-grained, is reported for systems with various size and oligomeric state. The performance was tested in the canonical- and replica exchange MD mode, with small- and moderate-size proteins and protein complexes (20 to 1,636 amino-acid residues), as well as with large systems such as, e.g., human proteosome 20S with size over 6,200 amino-acid residues, which show the advantage of using coarse-graining. It is demonstrated that, with using massively parallel architectures, and owing to the physics-based nature of UCGM, real-time simulations of the behavior of subcellular systems are feasible.
Keywords: Unified Coarse Grained Model, UNRES force field, molecular dynamics simula-tions, fine-grained parallelization, coarse-grained parallelization.
Introduction
The knowledge of the three-dimensional structure of macrobiomolecules is crucial for under-standing many biological processes. Most biologically important molecules are too large to handle for the classical simulation tools. Therefore, coarse-grained models and force fields have become useful in these studies [37]. Reduced representations of molecules allow saving computational cost of a few orders of magnitude in comparison with full-atomistic simulations. Various types of coarse-grained models such as the SICHO and CABS models have been developed [13, 14]. These knowledge-based protein models, developed in the Kolinski group, use only three degrees of freedom per residue. Compared to coarse-grained models of proteins [12], coarse-grained mod-els of polysaccharides have a relatively short history. Most of them use three or four interaction centers per sugar ring [26, 28, 34], including the polysaccharide component of the well-known MARTINI force field [24]. The MARTINI force field allows to simulate also other important biomolecules: lipids, proteins [29], DNA etc. [27]
The Unified Coarse Grained Model of biological macromolecules (UCGM) [19] that is be-ing developed in our laboratory is a physics-based model, designed to carry out large-scale simulations of biological macromolecules: proteins (the UNRES model [20]), nucleic acids (the NARES-2P model [7]), polysaccharides (the SUGRES-1P model), and lipid membranes. The chain representation in UCGM is reduced to two interaction sites (united peptide groups and united side chains in UNRES, united phosphate groups and united sugar-base groups in NARES-2P) or one interaction site (united sugar rings in SUGRES-1P). In UCGM the effective energy function was derived via generalized-cumulant expansion [17] of the potential of mean force of
1Institute of Informatics, Faculty of Mathematics, Physics and Informatics, University of Gda´nsk, Gda´nsk, Poland 2Faculty of Chemistry, University of Gda´nsk, Gda´nsk, Poland
biopolymer chains [21, 36]. Therefore, it reproduces the structure, dynamics, and thermody-namics of biopolymers very well despite the significant reduction of the number of interaction sites.
UNRES, which is the most advanced component of UCGM and has the longest history of development, has succeeded in physics-based protein-structure predictions [8, 15], investigation of protein-folding [38] and studying biological processes, such as amyloid formation [33], and iron-sulfur cluster biogenesis [30]. NARES-2P [7] can reproduce the structures and thermody-namics of small DNA and RNA molecules in free molecular-dythermody-namics simulations and has also recently been applied with success to model the stability of telomere sequences of DNA [35]. The preliminary version of SUGRES-1P [19] can reproduce the helical structure of amylose and the sheet-like structure of cellulose.
The article is organized as follows. Section 1 describes the UNRES model, force field, and coarse-grained molecular dynamics used in our study. In Section 2, we present details of imple-mentation of UNRES with Fortran 90. In Section 3, we report the results and their analysis. The Conclusion section summarizes our study.
1. Theory
1.1. UNRES Model and Force Field
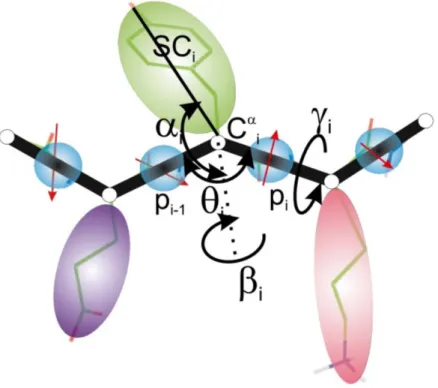
In the UNRES model (Fig. 1) [19], a polypeptide chain is represented as a sequence of α -carbon (Cα) atoms connected by virtual bonds, with united side chains (SC) attached to them (with different Cα· · ·SC bond lengths, dsc) and united peptide groups (p) positioned in the middle between the two consecutive Cαs. The SCs andps are the only interacting sites, while the Cαs only assist in geometry definition. Backbone geometry of the simplified polypeptide
chain is defined by the Cα· · ·Cα· · ·Cα virtual-bond angles θ (θ
i has the vertex at Cαi) and
the Cα· · ·Cα· · ·Cα· · ·Cα virtual-bond-dihedral angles γ (γ
i has the axis passing trough Cαi
and Cα
i+1). The local geometry of the ith side-chain center is defined by the zenith angle αi
(the angle between the bisector of the respective angle θi and the Cαi · · ·SCi vector) and the
azimuth angle βi (the angle of counter-clockwise rotation of the Cαi · · ·SCi vector about the
bisector from the Cα
i−1· · ·Cαi · · ·Cαi+1 plane, starting from Cαi−1). The energy function consists of local (including virtual-bond-deformation, virtual-bond-angle bending, virtual-bond-torsional and double torsional, and virtual-side-chain rotamer potentials), pairwise site-site interaction potentials, and correlation (multibody) potentials [20]. The solvent is implicit in the force field. UNRES was derived based on the potential of mean force (PMF) of polypeptide chains in water, which was factorized into Kubo cluster-cumulant functions [17], which are, in turn, identified with specific effective energy terms [21, 36]. Expansion of the cluster-cumulant functions into Kubo cluster cumulants [17] yielded approximate analytical expressions for the energy terms, including the correlation terms [21, 36]. The force field was calibrated with solution structural data of 7 traning proteins with various types of structure, by using the maximum-likelihood method recently developed in our laboratory [16].
Figure 1. UNRES model of polypeptide chains, with united peptide-groups (p)(blue circles), united side chains (SC)(ellipsoids) and α carbons (Cα)(small open circles)
1.2. Molecular Dynamics and Replica Exchange Molecular Dynamics with UNRES
Molecular dynamics is the core of the main conformational-search engine with UNRES. The equations of motion are Langevin equations because of implicit solvent treatment; however, for faster search, the simple Berendsen thermostat [2] (which originates from application of the Langevin equations to a system) is often used [9, 10]; the Nos´e–Hoover and Nos´e–Poincar´e thermostats that enable more strict control of temperature conservation were also implemented [11]. Because the virtual bonds are treated as stretchable rods, it was natural to select the Cα· · ·Cαand Cα· · ·SC virtual-bond vectors as variables. With such a representation, the inertia matrix is a full, albeit constant, matrix. Recently, by selecting the Cα and SC coordinates as variables, we reduced it to a pentadiagonal form (J. Sans and A. Liwo, to be submitted), this resulting in linear and not quadratic scaling the memory requirements with system size. A version of adaptive multiple-time-step velocity-Verlet algorithm was developed to integrate the equations of motion [9, 31].
MREMD method differs from REMD in that more than one trajectory is simulated at a given temperature.
2. Implementation
2.1. Parallelization
UNRES was parallelized on two levels: grained and fine-grained [23]. At the coarse-grained level, each MD trajectory is handled by a given task or task group. This means trivial parallelization scheme when many canonical MD trajectories are run, or task-farm-type par-allelization scheme for REMD/MREMD runs, with the master processor controlling the syn-chronization and exchange of replicas and the slave processors running only MD trajectories between the synchronization points. The REMD/MREMD UNRES jobs cannot be run in a serial mode, and the number of cores per job must be equal to the number of trajectories if coarse-grained-parallelization only is executed or a multiple of the number of trajectories for fine-grained parallelization. Because the management work is a small fraction of trajectory com-putations and the speed of calculations of all trajectories is virtually the same, the master processor also computes an MD trajectory. The efficiency of coarse-grained parallelization is close to 100 %.
To treat large systems, fine-grained parallelization was developed [23] in which energy and force evaluation was split between the tasks handling a given trajectory. The task-group master distributes coordinates to the slave tasks and collects the energy and force components from them; it also does its share of energy and force calculations.
2.2. Implementation with Fortran 90
Originally, UNRES was written in Fortran 77. Because of this, the code started to be more and more obscure as new functions were added to the package. Therefore, recently [25] we have rewritten the code in Fortran 90, grouping subroutines and data structures into clearly defined modules, which can also be shared by the programs for post-processing UNRES re-sults (WHAM, which executes the weighted-histogram analysis method [18] given the rere-sults of REMD/MREMD simulations in order to compute ensemble averages and probabilities of confor-mations and CLUSTER to group the conforconfor-mations into families [22]). This approach enabled us to reduce the redundancy in the code to a great extent. Moreover, the code takes advantage of dynamic memory allocation, this making memory usage more efficient, especially for 2-,3- and 4-dimensional arrays. The source code in the UNRES package with Fortran 90 has the following directory structure:
• unres (main program modules, source code with all UNRES functionalities);
– data (modules containing arrays declarations that correspond to COMMON block files in the UNRES package with Fortran 77);
• wham (weighted-histogram analysis method source code); • cluster (cluster analysis source code);
• xdrfpdb (format conversion programs source code).
3. Results and Discussion
We tested the scalability of the Fortran 90 implementation of UNRES with proteins of differ-ent size and on various hardware platforms, which included our local Beowulf cluster composed of 20-core Dell nodes Intel Xeon CPU E5-2640 v4 2.40 GHz connected with Gigabit Ethernet, at Faculty of Chemistry, University of Gda´nsk (piasek4.chem.univ.gda.pl), the 3214-node cluster, each node containing a 12-core Intel Xeon E5 v3 2.3 GHz processor (35,568 cores total), with Infiniband connection, at the Centre of Informatics - Tricity Academic Supercomputer & net-worK (CI TASK) in Gda´nsk (tryton-ap.gv.kdm.task.gda.pl), the 1084-node Cray supercomputer, each node containing 2 Intel Xeon E5-2690 v3 12-core processors with Cray Aries connection (okeanos.hpc.icm.edu.pl), and the 1024-node IBM Blue Gene/Q supercomputer, each node con-tainig 16 4-thread PowerPC A2 cores with 5D Torus connectivity (nostromo.hpc.icm.edu.pl), at the Interdisciplinary Centre for Mathematical and Computer Modelling, University of Warsaw. The proteins used for benchmark runs were as follows: tryptophan cage (20 residues, single chain, PDB code: 1L2Y), double-stranded RNA binding domain (68 residues, single chain, PDB code: 1STU), M-domain from the GAG polyprotein (87 residues, single chain, PDB code: 1A6S), endonuclease NucB (110 residues, single chain, PDB code: 5OMT), BID domain of Bep6 (137 residues, single chain, PDB code: 4YK1), CDI complex (263 residues, two chains, PDB code: 5HKQ), UDP-glucose glycoprotein glucosyltransferase (1,494 residues, single chain, PDB code: 5NV4), STRA6 receptor (819 residues, two chains, and 1,636 residues, four chains; PDB code: 5SY1) and human proteasome 20S (3,143 residues, 14 chains, and 6286 residues, 28 chains; PDB code: 4R3O).
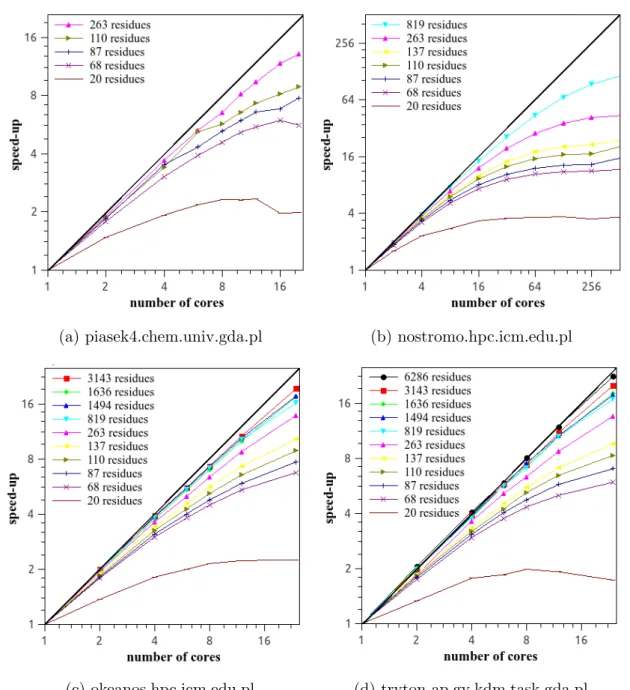
The speed-ups vs. the number of cores for single-trajectory MD runs are displayed in Fig. 2, while the corresponding efficiency plots are shown in Fig. 3. These data pertain to single-trajectory MD runs, i.e., they illustrate the performance of the software in the fine-grained parallel runs. As expected, the speed up and efficiency increase with the number of residues in a system. For the smallest protein, only up to 20-times speed up can be achieved, while for large proteins over 50 % efficiency can be achieved with up to 24 cores or up to 128 cores with Blue Gene/Q. Except for Blue Gene/Q, which has fast interconnect, all cores used for fine-grained parallelization were contained in a single node. Use of cores from more than one node for fine-grained parallelization resulted in performance deterioration. For example, use of 48 fine-grained processors (2 nodes) on the Cray resulted in the same or lower speed up than that with 24 processors (a single node).
The fraction of communication time split into synchronization (barrier) and data exchange is plotted for the 5HKQ protein in Fig. 4 as a function of the number of fine-grained tasks. It can be seen that communication constitutes less than 10 % of the total wall-clock time and that the main fraction of it is spent on synchronization, which is understandable because the data are exchanged between the cores of the same hardware unit.
fine-(a) piasek4.chem.univ.gda.pl (b) nostromo.hpc.icm.edu.pl
(c) okeanos.hpc.icm.edu.pl (d) tryton-ap.gv.kdm.task.gda.pl
Figure 2. Plots of the speed-ups for canonical single-trajectory UNRES/MD runs obtained for proteins with various chain length vs. the number of processors on various hardware platforms. The logarithmic scale with base 2 is used on both axes
grained tasks per trajectory. This is because all MD trajectories are running at virtually the same compute speed, this providing good load balancing and data transmission occurs rarely (in the reported calculations only 40 times).
cut-(a) piasek4.chem.univ.gda.pl (b) nostromo.hpc.icm.edu.pl
(c) okeanos.hpc.icm.edu.pl (d) tryton-ap.gv.kdm.task.gda.pl
Figure 3.Plots of the efficiencies for canonical single-trajectory UNRES/MD runs obtained for proteins with various numbers of residues vs. the number of processors on various hardware platforms. The logarithmic scale with base 2 is used on x-axis
Figure 4. Plots of the fraction of the collective-communication time to the non-setup wall-clock time with a given number of cores in a single-trajectory UNRES/MD run for the 5HKQ protein on okeanos.hpc.icm.edu.pl, tryton-ap.gv.kdm.task.gda.pl, piasek4.chem.univ.gda.pl and nostromo.hpc.icm.edu.pl. The right panel (zoom) shows a higher-magnification view of the bot-tom left region in the left panel. The logarithmic scale with base 2 is used on x-axis
(a) non-setup times (b) efficiencies
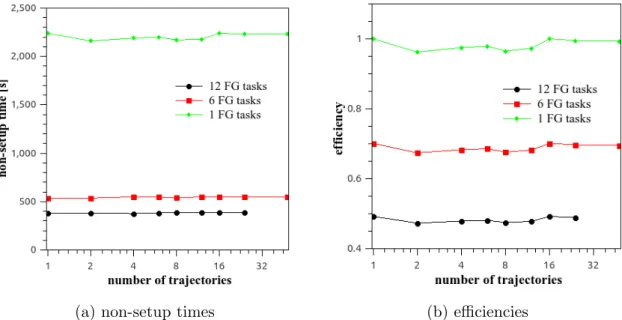
Figure 5. Plots of non-setup times and efficiencies in 40,0000 step MREMD simulations with replica exchange each 10,000 steps, numbers of trajectories from 1 (reference) to 48, and various numbers of fine-grain tasks per trajectory obtained for the 1A6S (87-residue) protein with tryton-ap.gv.kdm.task.gda.pl. The logarithmic scale with base 2 is used on x-axis. For a given point in the graph, the total number of cores used is obtained by multiplying the number of trajectories by the number of fine-grained tasks
Conclusion
Figure 6.Plots of non-setup times in 10,000 steps MD simulations with UNRES and GROMACS, with various numbers of processors obtained for the 1L2Y (20-residue), 1A6S (87-residue) and 5OMT (110-residue) proteins with tryton-ap.gv.kdm.task.gda.pl. The logarithmic scale with base 2 is used on both axes
tested its parallel efficiency both in the fine-grained mode (energy and force parallelization in single-trajectory molecular dynamics calculation) and in the full-blown (coarse- and fine-grained) mode, in which multiple trajectories were run in replica exchange calculations with communica-tion between the respective task groups every given number of steps, and parallelizacommunica-tion of the computation of each trajectory. For proteins with chain length about 300 amino-acid residues or more over 50 % parallel efficiency is obtained. The efficiency does not deteriorate when replica-exchange calculations are carried out.
The parallel implementation of UNRES enables us to simulate the dynamics of quite large proteins in real time. For example, for the 263-residue 5HKQ protein, the time required for 10,000 time steps with 24 fine-grained tasks run on the Cray machine is 24 seconds, which translates to about 176 nanoseconds of molecular-dynamics time per day (24 hours of computations) with the 4.89 fs time step commonly set in UNRES runs. However, because UNRES simulation time is at least 1,000-fold extended with respect to that of all-atom simulations with explicit solvent [9], one day of UNRES simulations for this protein amounts to a 0.176 millisecond molecular dynamics. With this speed up, physics-based simulations of biologically important processes are now at hand.
More informations about UNRES force field and the source code are available (De-scription of UNRES program. http://www.unres.pl/unres, accessed: 2018-06-15; Downloads page. http://unres.pl/downloads, accessed: 2018-06-15; UNRES server version. http://unres-server.chem.ug.edu.pl./about, accessed: 2018-06-15).
Acknowledgements
from the National Science Centre of Poland (Narodowe Centrum Nauki). Computational re-sources were provided by (a) the Interdisciplinary Center of Mathematical and Computer Mod-eling (ICM) at the University of Warsaw under Grant No. GA71-23, (b) the Centre of Informatics - Tricity Academic Supercomputer & networK (CI TASK) in Gda´nsk, and (c) our 796-processor Beowulf cluster at the Faculty of Chemistry, University of Gda´nsk.
This paper is distributed under the terms of the Creative Commons Attribution-Non Com-mercial 3.0 License which permits non-comCom-mercial use, reproduction and distribution of the work without further permission provided the original work is properly cited.
References
1. Abraham, M.J., Murtola, T., Schulz, R., P´all, S., Smith, J.C., Hess, B., Lindahl, E.: Gro-macs: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1-2, 19–25 (2015), DOI: 10.1016/j.softx.2015.06.001
2. Berendsen, H.J.C., Postma, J.P.M., van Gunsteren, W.F., DiNola, A., Haak, J.R.: Molecu-lar dynamics with coupling to an external bath. Journal of Chemical Physics 81(8), 3684– 3690 (1984), DOI: 10.1063/1.448118
3. Berendsen, H.J.C., van der Spoel, D., van Drunen, R.: Gromacs: A message-passing par-allel molecular dynamics implementation. Computer Physics Communications 91(1), 43–56 (1995), DOI: 10.1016/0010-4655(95)00042-E
4. Czaplewski, C., Kalinowski, S., Liwo, A., Scheraga, H.A.: Application of multiplexing replica exchange molecular dynamics method to the UNRES force field: Tests with α
and α+β proteins. Journal of Chemical Theory and Computation 5(3), 627–640 (2009), DOI: 10.1021/ct800397z
5. Duan, Y., Wu, C., Chowdhury, S., Lee, M.C., Xiong, G., Zhang, W., Yang, R., Cieplak, P., Luo, R., Lee, T., Caldwell, J., Wang, J., Kollman, P.: A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum me-chanical calculations. Journal of Computational Chemistry 24(16), 1999–2012 (2003), DOI: 10.1002/jcc.10349
6. Hansmann, U.H.E., Okamoto, Y.: Comparative study of multicanonical and simulated an-nealing algorithms in the protein folding problem. Physica A 212(3-4), 415–437 (1994), DOI: 10.1016/0378-4371(94)90342-5
7. He, Y., Maciejczyk, M., O ldziej, S., Scheraga, H.A., Liwo, A.: Mean-field interactions between nucleic-acid-base dipoles can drive the formation of a double helix. Physical Review Letters 110(9), 098101 (2013), DOI: 10.1103/PhysRevLett.110.098101
9. Khalili, M., Liwo, A., Jagielska, A., Scheraga, H.A.: Molecular dynamics with the united-residue model of polypeptide chains. II. Langevin and Berendsen-bath dynamics and tests on model α-helical systems. Journal of Physical Chemistry B 109(28), 13798–13810 (2005), DOI: 10.1021/jp058007w
10. Khalili, M., Liwo, A., Rakowski, F., Grochowski, P., Scheraga, H.A.: Molecular dynamics with the united-residue model of polypeptide chains. I. Lagrange equations of motion and tests of numerical stability in the microcanonical mode. Journal of Physical Chemistry B 109(28), 13785–13797 (2005), DOI: 10.1021/jp058008o
11. Kleinerman, D.S., Czaplewski, C., Liwo, A., Scheraga, H.A.: Implementations of Nos´e – Hoover and Nos´e –Poincar´e thermostats in mesoscopic dynamic simulations with the united-residue model of a polypeptide chain. Journal of Chemical Physics 128(24), 245103 (2008), DOI: 10.1063/1.2943146
12. Kmiecik, S., Gront, D., Kolinski, M., Wieteska, L., Dawid, A.E., Kolinski, A.: Coarse-grained protein models and their applications. Chemical Reviews 116(14), 7898–7936 (2016), DOI: 10.1021/acs.chemrev.6b00163
13. Kolinski, A., Skolnick, J.: Reduced models of proteins and their applications. Polymer 45(2), 511–524 (2004), DOI: 10.1016/j.polymer.2003.10.064
14. Kolinski, A., Skolnick, J.: Assembly of protein structure from sparse experimental data: An efficient Monte Carlo model. Proteins: Structure, Function, and Bioinformatics 32(4), 475–494 (1998), DOI: 10.1002/(SICI)1097-0134(19980901)32:43.0.CO;2-F
15. Krupa, P., Mozolewska, M.A., Wi´sniewska, M., Yin, Y., He, Y., Sieradzan, A.K., Ganzynkowicz, R., Lipska, A.G., Karczy´nska, A., ´Slusarz, M., ´Slusarz, R., Gie ldo´n, A., Czaplewski, C., Jagie la, D., Zaborowski, B., Scheraga, H.A., Liwo, A.: Performance of protein-structure predictions with the physics-based UNRES force field in CASP11. Bioin-formatics 32(21), 3270–3278 (2016), DOI: 10.1093/bioinBioin-formatics/btw404
16. Krupa, P., Ha labis, A., ˙Zmudzi´nska, W., O ldziej, S., Scheraga, H.A., Liwo, A.: Maxi-mum likelihood calibration of the UNRES force field for simulation of protein structure and dynamics. Journal of Chemical Information and Modeling 57(9), 2364–2377 (2017), DOI: 10.1021/acs.jcim.7b00254
17. Kubo, R.: Generalized cumulant expansion method. Journal of the Physical Society Japan 17(7), 1100–1120 (1962), DOI: 10.1143/JPSJ.17.1100
18. Kumar, S., Rosenberg, J.M., Bouzida, D., Swendsen, R.H., Kollman, P.A.: The weighted histogram analysis method for free-energy calculations on biomolecules. I. The Method. Journal of Computational Chemistry 13(8), 1011–1021 (1992), DOI: 10.1002/jcc.540130812
20. Liwo, A., Czaplewski, C., O ldziej, S., Rojas, A.V., Ka´zmierkiewicz, R., Makowski, M., Murarka, R.K., Scheraga, H.A.: Simulation of protein structure and dynamics with the coarse-grained UNRES force field. In: Voth, G. (ed.) Coarse-Graining of Con-densed Phase and Biomolecular Systems, chap. 8, pp. 1391–1411. CRC Press (2008), DOI: 10.1201/9781420059564.ch8
21. Liwo, A., Czaplewski, C., Pillardy, J., Scheraga, H.A.: Cumulant-based expressions for the multibody terms for the correlation between local and electrostatic interactions in the united-residue force field. Journal of Chemical Physics 115(5), 2323–2347 (2001), DOI: 10.1063/1.1383989
22. Liwo, A., Khalili, M., Czaplewski, C., Kalinowski, S., O ldziej, S., Wachucik, K., Scher-aga, H.A.: Modification and optimization of the united-residue (UNRES) potential energy function for canonical simulations. I. Temperature dependence of the effective energy func-tion and tests of the optimizafunc-tion method with single training proteins. Journal of Physical Chemistry B 111(1), 260–285 (2007), DOI: 10.1021/jp065380a
23. Liwo, A., O ldziej, S., Czaplewski, C., Kleinerman, D.S., Blood, P., Scheraga, H.A.: Imple-mentation of molecular dynamics and its extensions with the coarse-grained UNRES force field on massively parallel systems: Towards millisecond-scale simulations of protein struc-ture, dynamics, and thermodynamics. Journal of Chemical Theory and Computation 6(3), 583–595 (2010), DOI: 10.1021/ct9004068
24. Lopez, C.A., Rzepiela, A., de Vries, A.H., Dijkhuizen, L., Hunenberger, P.H., Marrink, S.J.: Martini coarse-grained force field: Extension to carbohydrates. Journal of Chemical Theory and Computation 5(12), 3195–3210 (2009), DOI: 10.1021/ct900313w
25. Lubecka, E.A., Liwo, A.: New UNRES force field package with FORTRAN 90. TASK Quarterly 20(4), 399–407 (2016), DOI: 10.17466/tq2016/20.4/n
26. Markutsya, S., Devarajan, A., Baluyut, J., Windus, T.L., Gordon, M.S., Lamm, M.H.: Evaluation of coarse-grained mapping schemes for polysaccharide chains in cellulose. Journal of Chemical Physics 138(21), 214108 (2013), DOI: 10.1063/1.4808025
27. Marrink, S.J., Tieleman, D.P.: Perspective on the Martini model. Chemical Society Reviews 42(16), 6801–6822 (2013), DOI: 10.1039/C3CS60093A
28. Molinero, V., Goddard III, W.A.: M3b: a coarse-grain force field for molecular simulations of malto-oligosaccharides and their water mixtures. Journal of Physical Chemistry B 108(4), 1414–1427 (2004), DOI: 10.1021/jp0354752
29. Monticelli, L., Kandasamy, S.K., Periole, X., Larson, R.G., Tieleman, D.P., Marrink, S.J.: The Martini coarse-grained force field: Extension to proteins. Journal of Chemical Theory and Computation 4(5), 819–834 (2008), DOI: 10.1021/ct700324x
31. Rakowski, F., Grochowski, P., Lesyng, B., Liwo, A., Scheraga, H.A.: Implementation of a symplectic multiple-time-step molecular dynamics algorithm, based on the united-residue mesoscopic potential energy function. Journal of Chemical Physics 125(20), 204107 (2006), DOI: 10.1063/1.2399526
32. Rhee, Y.M., Pande, V.S.: Multiplexed-replica exchange molecular dynamics method for protein folding simulation. Biophysical Journal 84(2), 775–786 (2003), DOI: 10.1016/S0006-3495(03)74897-8
33. Rojas, A., Liwo, A., Browne, D., Scheraga, H.A.: Mechanism of fiber assembly; treatment of Aβ-peptide peptide aggregation with a coarse-grained united-residue force field. Journal of Molecular Biology 404(3), 537–552 (2010), DOI: 10.1016/j.jmb.2010.09.057
34. Samsonov, S.A., Bichmann, L., Pisabarro, M.T.: Coarse-grained model of gly-cosoaminoglycans. Journal of Chemical Information and Modeling 55(1), 114–124 (2015), DOI: 10.1021/ci500669w
35. Sieradzan, A.K., Krupa, P., Wales, D.J.: What makes telomeres unique? Journal of Physical Chemistry B 121(10), 2207–2219 (2016), DOI: 10.1021/acs.jpcb.6b08780
36. Sieradzan, A.K., Makowski, M., Augustynowicz, A., Liwo, A.: A general method for the derivation of the functional forms of the effective energy terms in coarse-grained energy functions of polymers. I. Backbone potentials of coarse-grained polypeptide chains. Journal of Chemical Physics 146(12), 124106 (2017), DOI: 10.1063/1.4978680
37. Takada, S., Kanada, R., Tan, C., Terakawa, T., Li, W., Kenzaki, H.: Modeling structural dynamics of biomolecular complexes by coarse-grained molecular simulations. Accounts of Chemical Research 48(12), 3026–3035 (2015), DOI: 10.1021/acs.accounts.5b00338