A Haplotype-Based Algorithm for Multilocus Linkage Disequilibrium
Mapping of Quantitative Trait Loci With Epistasis
Xiang-Yang Lou,*
,†George Casella,* Ramon C. Littell,* Mark C. K. Yang,*
Julie A. Johnson
‡and Rongling Wu*
,1*Department of Statistics,‡Department of Pharmacy Practice, University of Florida, Gainesville, Florida 32611 and †Department of Agronomy, Zhejiang University, Hangzhou, Zhejiang 310029, People’s Republic of China
Manuscript received October 7, 2002 Accepted for publication January 16, 2003
ABSTRACT
For tightly linked loci, cosegregation may lead to nonrandom associations between alleles in a population. Because of its evolutionary relationship with linkage, this phenomenon is called linkage disequilibrium. Today, linkage disequilibrium-based mapping has become a major focus of recent genome research into mapping complex traits. In this article, we present a new statistical method for mapping quantitative trait loci (QTL) of additive, dominant, and epistatic effects in equilibrium natural populations. Our method is based on haplotype analysis of multilocus linkage disequilibrium and exhibits two significant advantages over current disequilibrium mapping methods. First, we have derived closed-form solutions for estimating the marker-QTL haplotype frequencies within the maximum-likelihood framework implemented by the EM algorithm. The allele frequencies of putative QTL and their linkage disequilibria with the markers are estimated by solving a system of regular equations. This procedure has significantly improved the computational efficiency and the precision of parameter estimation. Second, our method can detect marker-QTL disequilibria of different orders and QTL epistatic interactions of various kinds on the basis of a multilocus analysis. This can not only enhance the precision of parameter estimation, but also make it possible to perform whole-genome association studies. We carried out extensive simulation studies to examine the robustness and statistical performance of our method. The application of the new method was validated using a case study from humans, in which we successfully detected significant QTL affecting human body heights. Finally, we discuss the implications of our method for genome projects and its extension to a broader circumstance. The computer program for the method proposed in this article is
available at the webpage http://www.ifasstat.ufl.edu/genome/ⵑLD.
D
ESPITE its significant importance in plant and ani- occur in a population. Linkage and nonrandomassocia-tion leading to cosegregaassocia-tion of different genes affect mal breeding and evolutionary studies, the genetic
basis of a quantitatively inherited trait has not been gamete distribution and frequencies and, thus, are
con-sidered to be population genetic parameters (Mackay
well understood. Two factors have caused this. First,
a quantitative trait, or a complex trait, is likely to be 2001).
controlled by many genes, each having a small effect Apart from possible allelic associations, different
(LynchandWalsh1998). Such genes are difficult to genes may be mutually dependent when one gene
pre-identify using classic genetic approaches. Second, as vents another from manifesting its effect through a
par-revealed in a considerable body of literature, the genes ticular biochemical pathway (Phillips1998). Such
de-underlying a complex trait are not independent in ex- pendence of genes is referred to asepistasis, a ubiquitous
erting an effect on quantitative variation (reviewed in phenomenon in the genetic control of a complex trait.
Mackay2001). The dependence between alleles at dif- Epistasis, described as a type of gene interaction on
ferent loci may be due to their linkage on the same phenotypes, possesses the property of a quantitative
ge-chromosome or nonrandom association even if they are netic parameter. Classical estimation of quantitative
ge-on different chromosomes. In particular, the nge-onran- netic parameters is based on the resemblance among
dom association between two linked genes is calledlink- relatives using a theory established byFisher (1918),
age (gametic) disequilibrium (Risch and Merikangas but this approach lacks the power to estimate epistasis
1996;Hudson2000). Unlinked genes may also be asso- because epistasis contributes little to the relative
resem-ciated if biological processes, such as population differ- blance. A lack of powerful methods for estimating
epista-entiation, population admixture, and natural selection, sis has diminished its central role in trait evolution and
domestication, as claimed by S. Wright and his followers
(Wolfet al.2000).
The advent of complete DNA-marker linkage maps
1Corresponding author:Department of Statistics, 533 McCarty Hall C,
University of Florida, Gainesville, FL 32611. E-mail: [email protected] has provided a powerful tool for investigating the
netic architecture of a quantitative trait in plants, ani- natural population. Our method has been validated by the successful detection of QTL for human body height.
mals, or humans (reviewed in Mackay2001; Barton
andKeightley2002). A number of statistical methods have been proposed to map genes responsible for
com-POPULATION GENETIC MODEL
plex traits, referred to as quantitative trait loci (QTL),
based on different genetic designs, different marker Suppose there is a panmictic natural population, in
types, and different mapping populations (reviewed in whichmbiallelic markers,ᏹ
1,ᏹ2, . . . ,ᏹm, andqbiallelic Jansen2000;Hoeschele2000). Most of these methods QTL, ᏽ1,ᏽ2, . . . ᏽq, are segregating. For the sake of
were developed to adapt gene segregation patterns in distinction, we use the subscripts to denote the markers
a mapping pedigree derived from a controlled cross and the superscripts to denote the QTL. Two alleles at
and have two significant limits when they are applied a locus are denoted by M1
k and M2k, or indexed by rk
on a wider scheme. First, for many species such as hu- (r
k⫽ 1, 2), for markerᏹk, andQl1andQl2, or indexed
mans, it is not possible to obtain controlled crosses and bys
l(sl⫽1, 2), for QTLᏽl. This population is assumed
thus their QTL mapping should be based on existing to be in disequilibrium, so that all markers and QTL
natural populations. Second, many methods assume are associated with one another. We denoteD
k1k2as the
that epistatic effects due to gene interaction are absent, coefficient of digenic linkage disequilibrium between
to facilitate the estimation of quantitative genetic pa- markersᏹk
1 and ᏹk2, D
l1l2 as the coefficient of digenic
rameters. Given the complexity of quantitative variation, linkage disequilibrium between QTL ᏽl1 and ᏽl2 and
however, the assumption of no epistasis most likely devi- Dl1
k1as the coefficient of digenic linkage disequilibrium
ates from biological reality (Mackay2001;Bartonand between markerᏹk
1and QTLᏽ
l1. Similarly, the
coeffi-Keightley2002). cients of trigenic (Dl1
k1k2andD l1l2
k1), quadrigenic (D
l1l2 k1k2), or
In this article, we present a new statistical method for higher-order linkage disequilibrium can be defined.
mapping QTL with epistasis in natural populations. Our Haplotypes, zygote configurations, and zygote
geno-analysis is based on an important population property types:With the above notation for the markers and QTL
of gene segregation, that is, linkage disequilibrium and their corresponding alleles, a general multilocus
(LynchandWalsh1998;Hudson2000), although this haplotypemix for the markers (M1 M2. . .M
m) and the
property made quantitative genetic study more difficult QTL (Q1Q2. . .Qq) is denoted bygs1s2...sq
r1r2...rm(r1, . . . ,rm;s1,
in the past. Linkage causing linkage disequilibrium is . . . , s
q⫽ 1, 2), with population frequency expressed
used as a basis for fine mapping of human diseases asps1s2...sq
r1r2...rm. This haplotype frequency can be divided into
(Templeton1999) and has been successful in several the allele frequencies and a set of disequilibrium
coeffi-case studies of gene positional cloning (reviewed in cients of different orders (Hudson2000). For example,
Ardlie et al.2002).Luo andSuhai(1999), Luoet al. when there is no interference between recombinations,
(2000), andWuet al.(2002) extended disequilibrium- a one-marker (ᏹk)/one-QTL (ᏽl) haplotype frequency
based mapping strategies to map QTL for a quantitative is divided into two parts, no disequilibrium and digenic
trait using a random sample drawn from a natural popu- disequilibrium, expressed as
lation. However, in these studies only the simplest
model, based on one marker and one QTL, was formu- No disequilibrium psl
rk⫽ prkpsl
Digenic disequilibrium ⫹(⫺1)rk⫹slDl
k.
(1) lated. The application of these models to study the
ge-netic architecture of polygenic traits is therefore limited.
Our model presented here uses a multilocus linkage From now on we omit the symbolskfor markers andl
disequilibrium analysis to detect epistatic QTL and esti- for QTL for simplicity, unless they are needed. A
two-mate the effects of their gene action and interaction on marker/one-QTL haplotype frequency is expressed as
a quantitative trait. Our model can be considered to be comprehensive, because it includes all possible disequi-libria among different markers and QTL and considers
No disequilibrium ps
r1r2⫽pr1p
sp r2
3 digenic disequilibria ⫹(⫺1)s⫹r2pr
1D
l k2⫹(⫺1)
r1⫹spr 2D
l k1
⫹(⫺1)r1⫹r2psDk 1k2
1 trigenic disequilibrium ⫺(⫺1)r1⫹r2⫹sDlk
1k2.
quantitative gene interaction effects of different kinds.
We derive a simple closed form of the expectation-max- (2)
imization (EM) algorithm for estimating the allelic fre- When four or more genetic loci are associated,
disequi-quencies and coefficients of linkage disequilibria and libria of one set of loci may interact with those of other
gene action and interaction effects. Because of this com- sets to form so-called multiplicative disequilibria (
Ben-putational innovation, the estimation precision of QTL nett1954). When (at least two) epistatic QTL are
asso-population and quantitative parameters from our ciated with multiple markers, these multiplicative
dis-multilocus model can be significantly improved over equilibria should be considered. For example, under
that of traditional treatments, despite the fact that the the assumption of no interference between
recombina-multilocus model contains an increasingly larger num- tion events (Bennett 1954), a four-marker/two-QTL
ber of unknown parameters. Extensive simulations are haplotype frequency is expressed as
performed to study the robustness and performance of
ps1s2
r1r2r3r4⫽
No disequilibrium different alleles at the same locus, the number of zygote configurations may be greater than the number of zy-pr1pr2pr3pr4p
s1ps2
gote genotypes. For example, zygote genotypeG{12}
{11}{12}is
15 digenic disequilibria
generated via configurationg1
11⫻g212org211⫻ g112.
⫹(⫺1)r3⫹r4pr 1pr2p
s1ps2Dk
3k4⫹. . .⫹(⫺1)
s1⫹s2pr 1pr2pr3pr4D
l1l2
Of the eight three-locus haplotype frequencies
con-20 trigenic disequilibria
tained in the joint probability matrix of Table 1, seven
⫺(⫺1)r2⫹r3⫹r4pr 1p
s1ps2Dk
2k3k4⫺. . .⫺(⫺1)
r1⫹s1⫹s2pr 2pr3pr4D
l1l2
k1
are independent because of the constraint 兺2
r1⫽1兺r22⫽1
15 quadrigenic disequilibria
兺2
s⫽1psr1r2⫽1. These seven independent frequencies,
ar-⫹(⫺1)r1⫹r2⫹r3⫹r4ps1ps2Dk
1k2k3k4⫹. . .⫹(⫺1)
r1⫹r2⫹s1⫹s2pr 3pr4D
l1l2
k1k2 rayed by⍀
p⫽ (psr1r2)1⫻7, are composed of three allele
6 pentagenic disequilibria frequencies (p
r1,pr2,ps), three digenic linkage
disequilib-⫺(⫺1)r1⫹r2⫹r3⫹r4⫹s2ps1Dl2
k1k2k3k4⫺. . .⫺(⫺1)
r1⫹r2⫹r3⫹s1⫹s2p
r4D
l1l2
k1k2k3 ria (Dk
1k2,D
l
k1,Dlk2), and one trigenic linkage
disequilib-1 hexagenic disequilibrium rium (Dl
k1k2). We develop a statistical algorithm to
esti-⫹(⫺1)r1⫹r2⫹r3⫹r4⫹s1⫹s2Dl1l2
k1k2k3k4 ate⍀pand ultimately estimate those population genetic
45 digenic⫻digenic disequilibria parameters describing allelic association and
popula-tion evolupopula-tionary history.
⫹(⫺1)r1⫹r2⫹r3⫹r4ps1ps2Dk
1k2Dk3k4⫹. . .
A more complicated situation is that in which two
60 digenic⫻trigenic disequilibria
QTL (ᏽl1andᏽl2) are detected by two pairs of markers
⫺(⫺1)r1⫹r2⫹r3⫹r4⫹s1ps2Dk 1k2D
l1
k3k4⫺. . .
(ᏹk1 ⫺ᏹk2andᏹk3 ⫺ᏹk4). We permit these six loci to
15 digenic⫻quadrigenic disequilibria
be mutually associated in the population. A total of 64
⫹(⫺1)r1⫹r2⫹r3⫹r4⫹s1⫹s2Dk 1k2D
l1l2
k3k4⫹. . .
(26) six-locus haplotypes randomly unite to generate
10 trigenic⫻trigenic disequilibria
729 (36) six-locus zygotes whose population frequencies
⫹(⫺1)r1⫹r2⫹r3⫹r4⫹s1⫹s2Dk 1k2k3D
l1l2
k4 ⫹. . . are arrayed by matrix Pl1l2
k1k2k3k4⫽ {P {s1s⬘1}{s2s⬘2}
{r1r⬘1}{r2r⬘2}{r3r⬘3}{r4r⬘4}}(81⫻9). If
15 digenic⫻digenic⫻digenic disequilibria these six loci contain two independent sets each with
⫹(⫺1)r1⫹r2⫹r3⫹r4⫹s1⫹s2D
k1k2Dk3k4D
s1s2⫹. . . .
two markers and one QTL, we have Pl1l2
k1k2k3k4 ⫽P l1 k1k2 丢
(3)
Pl2
k3k4, where丢 is the Krockner product. If the two sets
For themmarkers andqQTL, a total of 2m⫹qhaplotypes
are not independent, however, the joint zygote
probabil-randomly unite to generate 3m⫹q zygote genotypes
con-ities of four markers and two QTL should be derived on
taining 2m⫹q(2m⫹q⫹1)/2zygote configurationsor modes of
the basis of different zygote configurations via random zygote formation via different combinations of paternal
combinations of the 64 haplotypes. Summing to 1, the and maternal gametes. The number of zygote genotypes
64 six-locus haplotype frequencies contain 63 indepen-is smaller than that of zygote configurations because
dent frequencies (arrayed as⍀p), which are expressed
different configurations may generate the same zygote
in terms of an equal number of composite parameters, genotype. A zygote genotype can be generally
formu-six-allele frequencies, and 57 coefficients of linkage dis-lated as
equilibria of different orders. G{s1s⬘1}{s2s⬘2}. . .{sqs⬘q}
{r1r⬘1}{r2r2⬘}. . .{rmr⬘m}, Multiallelic disequilibrium analysis: For outcrossing
populations, it is not uncommon to have multiple alleles
where {·}{·} are used to separate two different loci, and at a locus. Although the measure of linkage
disequilib-r1ⱕr⬘1, . . . ,rmⱕr⬘m,s1ⱕs⬘1, . . . ,sqⱕs⬘q ⫽1, 2 are the rium between two multiallelic loci was well defined
two alternative alleles of a zygote at the same locus. The (Weir1996), a multiallelic disequilibrium analysis has
joint frequency of a zygote for all markers and QTL is not been extended to many different loci, because of
expressed as an increasingly large number of disequilibrium
coeffi-cients. Assume that there areukalleles for markerᏹkand
P{s1s⬘1}{s2s⬘2}. . .{sqs⬘q}
{r1r⬘1}{r2r⬘2}. . .{rmr⬘m}. v
lfor QTL ᏽl. According toBennett (1954), digenic
linkage disequilibrium between marker allele rk and
At Hardy-Weinberg equilibrium, the population
fre-quency of a (m⫹q)-locus zygotic configuration is calcu- QTL alleleslcan be defined as
lated as the product of the two corresponding haplotype
Dsl rk⫽p
sl rk⫺prkp
sl
frequencies (LynchandWalsh1998). Yet, the
popula-tion frequency of a (m⫹q)-locus zygotic genotype is the with constraints兺uk
rk⫽1D
sl rk⫽兺
vl sl⫽1D
sl
rk⫽0 . Trigenic
link-summation of the population frequencies of all possible age disequilibrium among two marker alleles r
k1 and
zygotic configurations. r
k2at markersᏹk1 andᏹk2and one QTL alleleslat QTL
Table 1 describes a matrix for the population
frequen-ᏽlis expressed as
cies of 27 zygote genotypes at two markers and one QTL, which contain 36 zygote configurations through random combinations of the eight haplotypes. As shown
Haplotype frequency Dsl
rk1rk2⫽p
sl
rk1rk2
3 disequilibria ⫺pslD
rk1rk2⫺prk2D
sl
rk1⫺prk1D
sl
rk2
No disequilibrium ⫺prk1prk2p
sl,
in the table, we use P( ⫽ 1, . . . , 36) to denote the
frequencies of different zygote configurations. Because different configurations may produce the same zygote
genotype when paternal and maternal gametes have with constraints兺uk1
rk 1⫽1
Dsl
rk 1rk2⫽兺
uk2
rk 2⫽1
Dsl
rk1rk2⫽兺 vl sl⫽1D
sl rk1rk2⫽
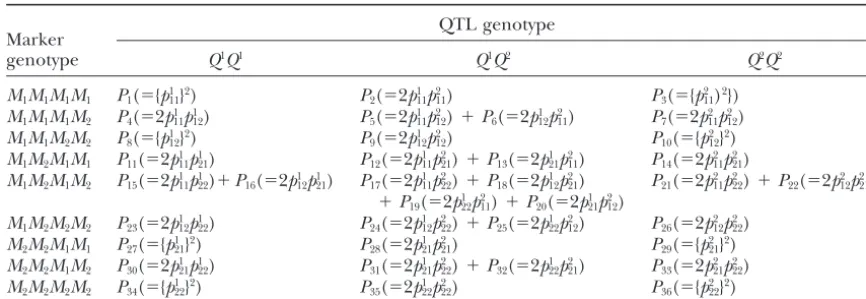
TABLE 1
Joint zygote probabilities of the QTL genotypes and two-marker genotypes in terms of
zygote configurationsP(⫽1, . . . , 36)
QTL genotype Marker
genotype Q1Q1 Q1Q2 Q2Q2
M1M1M1M1 P1(⫽{p111}2) P2(⫽2p111p211) P3(⫽{p211)2}) M1M1M1M2 P4(⫽2p111p112) P5(⫽2p111p212)⫹P6(⫽2p112p112) P7(⫽2p211p212) M1M1M2M2 P8(⫽{p112}2) P9(⫽2p112p212) P10(⫽{p212}2) M1M2M1M1 P11(⫽2p111p121) P12(⫽2p111p221)⫹P13(⫽2p121p112) P14(⫽2p211p221)
M1M2M1M2 P15(⫽2p111p122)⫹P16(⫽2p112p211) P17(⫽2p111p222)⫹P18(⫽2p112p221) P21(⫽2p211p222)⫹P22(⫽2p212p221) ⫹P19(⫽2p122p211)⫹P20(⫽2p121p212)
M1M2M2M2 P23(⫽2p112p122) P24(⫽2p112p222)⫹P25(⫽2p122p122) P26(⫽2p212p222) M2M2M1M1 P27(⫽{p121}2) P28(⫽2p121p221) P29(⫽{p221}2) M2M2M1M2 P30(⫽2p121p122) P31(⫽2p121p222)⫹P32(⫽2p122p212) P33(⫽2p221p222) M2M2M2M2 P34(⫽{p122}2) P35(⫽2p122p222) P36(⫽{p222}2)
The expression for the frequency of each zygote configuration due to random recombination of paternal and maternal gametes is given in parentheses (see the text).
probabilities of the QTL genotypes and the marker ge-Hexagenic linkage disequilibria between four marker
notypes on the basis of their zygote formation. We now nonalleles and two QTL nonalleles are expressed as
introduce a genetic model for characterizing the addi-Dsl1sl2
rk 1rk2rk3rk3
⫽ tive, dominant, and epistatic effects of QTL affecting a
quantitative trait. We useMatherandJinks’(1982)
nota-Haplotype frequency
tion to describe the epistasis, under which the genotypic psl1sl2
rk
1rk2rk3rk4 values for any two QTL (ᏽl1andᏽl2) are expressed as
15 digenic disequilibria
⫺prk3prk4p sl1psl2Drk
1rk2⫺ . . .⫺prk1prk2prk3prk4D sl1sl2
20 trigenic disequilibria
⫺prk4p sl1psl2Dr
k1rk2rk3⫺. . .⫺prk1prk2prk3D sl
1sl2 rk
4
Ql1genotype
{s1s⬘
1}{s2s⬘2}
Q1Q1 Q1Q2 Q2Q2
Ql 2genotype
Q1Q1 Q1Q2 Q2Q2 ⫹al1⫹al2⫹il1l2 ⫹al1⫹dl2⫹jl1l2 ⫹al1⫺al2⫺il1l2
⫹dl
1⫹al2⫹kl1l2 ⫹dl1⫹dl2⫹ll1l2 ⫹dl1⫺al2⫺kl1l2
⫺al1⫹al2⫺il1l2 ⫺al1⫹dl2⫺jl1l2 ⫺al1⫺al2⫹il1l2
s1ⱕs⬘1,s2ⱕs2⬘⫽1, 2
, 15 quadrigenic disequilibria
⫺psl1psl2Dr
k1rk2rk3rk4⫺. . .⫺prk1prk2D sl1sl2 rk3rk4
6 pentagenic disequilibria
(4)
⫽
⫺psl2Dsl1 rk1rk2rk3rk4⫺
. . .⫺prk1D sl1sl2
rk2rk3rk4 where is the overall mean; al1,al2,dl1, anddl2 are the
45 digenic⫻digenic disequilibria additive and dominant effects of the two QTL, respectively;
andil1l2,jl1l2,kl1l2, andll1l2 are the additive⫻additive,
addi-⫺prk3prk4p
sl1psl2Drk1rk2⫺. . .⫺prk1prk2prk3prk4D sl1sl2
tive⫻dominant, dominant⫻additive, and dominant⫻
60 digenic⫻trigenic disequilibria
dominant epistatic effects between the two QTL,
respec-⫺prk 1Drk2rk3D
sl 1sl2 rk
4
⫺. . .⫺psl 2D
sl 1 rk 4
Drk
1rk2rk3 tively.
15 digenic⫻quadrigenic disequilibria The phenotypic value of a quantitative trait due to these
⫺Drk1rk2rk3rk4D sl
1sl2⫺. . .⫺Drk1rk2D sl
1sl2
rk3rk4 two putative QTL for individualifrom a random sample
of the population can be written as
10 trigenic⫻trigenic disequilibria
⫺Drk1rk2rk3D sl1sl2 rk4 ⫺
. . .⫺Dsl1sl2 rk1
Drk2rk3rk4
yi⫽
兺
s1ⱕs⬘1 s2ⱕ
兺
s⬘2{s1s⬘1}{s2s⬘2}x
iS1S⬘1S2S⬘2⫹ei, (5)
15 digenic⫻digenic⫻digenic disequilibria
⫺Drk1rk2Drk3rk4D
sl1sl2⫺. . .⫺Dsl2 rk1
Dsl1 rk4
Drk2rk3
wherexiS1S1⬘S2S⬘2is an indicator variable, defined as 1 if
individ-No disequilibrium
ualihas QTL genotypeQs1Qs1⬘Qs2Qs2⬘and 0 otherwise, and
⫺prk1prk2prk3prk4p sl1psl2,
eiis the residual error, distributed asN(0,2). It is
possi-ble that each QTL genotype has a different residual
with constraints兺uk1
rk 1⫽1
Dsl1sl2 rk
1rk2rk3rk4
⫽. . .⫽兺vl2 sl
2⫽1
Dsl1sl2 rk
1rk2rk3rk4
⫽0.
variance. Since there are nine QTL genotypes, nine independent genotypic values arrayed by {{s1s⬘1}{s2s⬘2}}
1⫻9can
be defined as in (4). These nine genotypic values are QUANTITATIVE GENETIC MODEL
composed of nine unknown quantitative genetic
param-In the above section, we discussed the population eters: one overall mean (), two additive {al1,al2}, two
achieved by differentiating the log-likelihood with
re-kl1l2,ll1l2}. The nine genotypic values and residual variance
spect to⍀, which leads to
comprise a quantitative genetic parameter vector
ar-rayed by ⍀q ⫽ {{11}{11}, . . . , {22}{22}, 2}, which can be
⍀logL(y,M,g|⍀)⫽r1兺ⱕr⬘r2兺ⱕr⬘
2
冦
⫽兺361
z{r1r⬘
1}{r2r⬘2} ·
⍀P
logP
estimated by incorporating the population genetic properties of the QTL within a mapping framework.
For many outcrossing populations, the biallelic-QTL
⫹ 兺
n{r1r⬘1}{r2r⬘2} i⫽1 兺
36
⫽1
z{r1r⬘
1}{r2r⬘2}i兺 sⱕs⬘
x{ss⬘}
⍀q
logf{ss⬘}(yi)
冧
, genetic model described by Equation 5 may not besuf-ficient and should be extended to a multiallelic-QTL
(8) situation. Quantitative genetic models based on
multial-lelic inheritance have been well discussed in the litera- where z
{r1r⬘1}{r2r⬘2} ·⫽ 兺 n{r
1r⬘1}{r2r⬘2}
i⫽1 z{r1r⬘1}{r2r⬘2}i. By letting the score
ture (seeCockerham1980).
(Equation 8) equal zero and solving the log-likelihood equation, we obtain the maximum-likelihood estimates (MLE) of the unknown parameters as
STATISTICAL MODEL
Likelihood of the complete data: In this QTL map- pˆ1
11⫽
1
2n(2n1⫹n2⫹n4⫹n5⫹n11⫹n12⫹n15⫹n17)
ping study, marker data (M) and phenotype data (y)
for all n individuals randomly drawn from a natural
pˆ2 11⫽
1
2n(n2⫹2n3⫹n6⫹n7⫹n13⫹n14⫹n19⫹n21)
population are observed data, whereas zygote formation
modes,i.e.,zygote configurations (g), are unobservable
or missing data. The observed data alone are incomplete
pˆ1 12⫽
1
2n(n4⫹n6⫹2n8⫹n9⫹n16⫹n18⫹n23⫹n24)
data, which, along with the missing data, comprise the complete data. For simplicity, the statistical framework
is illustrated for the relatively simple two-marker/one- pˆ2
12⫽
1
2n(n5⫹n7⫹n9⫹2n10⫹n20⫹n22⫹n25⫹n26)
QTL model. This framework can be extended to consider a multiple-marker/multiple-QTL model, considering
pˆ1 21⫽
1
2n(n11⫹n13⫹n16⫹n20⫹2n27⫹n28⫹n30⫹n31)
multiple alleles at each locus. The likelihood function of the complete data given the unknown parameters
⍀ ⫽ (⍀p, ⍀q) is written, under the two-marker/one- pˆ2
21⫽
1
2n(n12⫹n14⫹n18⫹n22⫹n28⫹2n29⫹n32⫹n33)
QTL model, as
pˆ1 22⫽
1
2n(n15⫹n19⫹n23⫹n25⫹n30⫹n32⫹2n34⫹n35)
L(y,M,g|⍀)⫽
兿
r1ⱕr⬘1
兿
r2ⱕr⬘2
兿
n{r1r⬘1}{r 2r⬘2}
i⫽1
pˆ2 22⫽
1
2n(n17⫹n21⫹n24⫹n26⫹n31⫹n33⫹n35⫹2n36)
⫻
冤
兺
36⫽1
z{r1r⬘1}{r2r⬘2}iP
兺
sⱕs⬘x{ss⬘}
f{ss⬘}(yi)
冥
, (6){ss⬘}⫽
兺
r1ⱕr⬘1兺
r2ⱕr⬘2兺
n{r 1r⬘1}{r2r⬘2}
i⫽1
兺
36⫽1z{r1r⬘1}{r2r⬘2}ix
{ss⬘} yi
兺
r1ⱕr⬘1兺
r2ⱕr⬘2兺
n{r 1r⬘1}{r2r⬘2}
i⫽1
兺
36⫽1z{r1r⬘1}{r2r⬘2}ix
{ss⬘}
or
L(y,M,g|⍀)⫽
兿
r1ⱕr⬘1
兿
r2ⱕr⬘2
兿
n{r1r1}{⬘r2r⬘2}
i⫽1
兿
36⫽1
2⫽1
nr
兺
1ⱕr⬘1
兺
r2ⱕr⬘2
兺
n{r1r⬘
1}{r2r⬘2}
i⫽1
兺
36⫽1
z{r1r⬘1}{r2r⬘2}i
兺
sⱕs⬘
x{ss⬘}
(yi⫺ ˆ{ss⬘})2,
⫻
冤
P兺
sⱕs⬘
x{ss⬘} f{ss⬘}(yi)
冥
z{r 1r⬘1}{r2r⬘2}i
, (7)
wheren⫽ 兺r1ⱕr⬘1 兺r2ⱕr2⬘z{r1r⬘1}{r2r⬘2}·denotes the sample size
of a particular zygote configuration( ⫽1, . . . , 36)
as defined in Table 1. For the complete data,is
observ-wheren{r1r⬘1}{r2r⬘2}is the observed number of individuals with
able. Based on the invariance property of maximum-marker genotypeMr1Mr⬘1Mr2Mr⬘2,
likelihood estimates, the MLEs of allelic frequencies, the coefficients of linkage disequilibrium, and QTL ef-z{r1r⬘1}{r2r⬘2}i⫽
冦
1 if individualiwithin incomplete category Mr1Mr⬘1Mr2Mr⬘2
belongs to complete category( ⫽1, . . . , 36)
0 otherwise
fects can be obtained from
aˆ⫽1
2(
{11}⫺ {22})
x{ss⬘}
⫽
冦
1 if the QTL genotype of zygote formationis from
categoryQsQs⬘(僆{QsQs⬘})
0 otherwise, dˆ⫽ {12}⫺ 1
2(
{11}⫹ {22})
andf{ss⬘}(yi) is a normal density of observationyi with a pˆ1(k
1)⫽pˆ
1
11⫹pˆ112 ⫹pˆ112⫹ pˆ212
particular QTL genotype QsQs⬘having mean {ss⬘} and
pˆ1(k2)⫽pˆ111⫹pˆ112 ⫹pˆ121⫹ pˆ221
qˆ1 ⫽pˆ1
11⫹pˆ112⫹ pˆ121⫹pˆ122 ⫻
冤
⍀p
logP⫹
兺
sⱕs⬘ xss⬘
⍀q
logf{ss⬘}(yi)
冥
⌸{r1r⬘1}{r2r⬘2}i,Dˆl
k1 ⫽pˆ111⫹ pˆ121 ⫺pˆ1(k1)qˆ1 (10)
Dˆl
k2 ⫽pˆ111⫹ pˆ211 ⫺qˆ1pˆ1(k2) where
Dˆk1k2 ⫽pˆ111⫹ pˆ211⫺pˆ1(k1)pˆ1(k2)
⌸{r1r⬘1}{r2r⬘2}i⫽
w{r1r⬘1}{r2r⬘2}P
兺
sⱕs⬘x ss⬘ f{ss⬘}(yi)
兺
36⫽1
关
w{r1r⬘1}{r2r⬘2}P兺
sⱕs⬘x ss⬘ f{ss⬘}(yi)
兴
, Dˆl
k1k2⫽pˆ 1
11⫺ pˆ1(k1)Dˆlk2 ⫺pˆ1(k2)Dˆlk1⫺qˆ1Dˆk1k2 ⫺pˆ1(k1)qˆ1pˆ1(k2).
(11)
The likelihood of incomplete data:The data of QTL
mapping are incomplete because only marker data and
which could be thought of as a posterior probability that phenotype data can be observed and the data on zygote
individualihas a marker-QTL zygote configurationgiven
configurations and QTL genotypes are missing. The
marker zygote genotype G{r1r⬘1}{r2r2⬘}. We then implement
likelihood of incomplete data is built upon a mixture
the EM algorithm (Dempster et al. 1977) with the
ex-model in that each individual is assumed to have arisen
panded parameter set {⍀, ⌸}, where⌸⫽{⌸{r1r⬘1}{r2r2⬘}i,i⫽
from one of these unknown QTL genotypes. Finite
mix-1, . . . ,n{r1r⬘1}{r2r⬘2};r1ⱕr⬘1,r2ⱕr2⬘⫽1, 2}.
tures of distributions used to model a wide variety of
Each iteration consists of two steps. First, on the E
random phenomena (seeMcLachlanandPeel2000)
step, the complete-data log-likelihood is averaged over can provide a sound mathematical-based approach for
the conditional distribution of the indicator variables distinguishing unknown QTL genotypes. With a normal
given the observed data, using the current estimate of mixture model-based approach for separating QTL
ge-the parameter vector. Since ge-the complete-data log-likeli-notypes, the likelihood function of incomplete data
in-hood is linear in these indicator variables, the E-step of
cluding the phenotype (y) and marker information (M)
is written as the EM algorithm involves simply replacing them by the
current values of their conditional expectations, that is, L(y,M|⍀)⫽
兿
r1ⱕr⬘1
兿
r2ⱕr⬘2兿
n{r1r1⬘}{r2r⬘2}i⫽1
the posterior probabilities of component membership expressed in Equation 11. Next, in the M step,
condi-tional on⌸, we solve for the zeros of Equation 10
(likeli-⫻
冦
Prob(G{r1r1⬘}{r2r⬘2})兺
36⫽1
冤
P|{r1r⬘1}{r2r⬘2}
兺
sⱕs⬘
x{ss⬘}f{ss⬘}(yi)
冥冧
,hood equations) to get our estimates of⍀. The
likeli-hood equation can be split into two terms: The first term refers to the genetic association as specified by
⫽
兿
r1ⱕr⬘1
兿
r2ⱕr2⬘兿
n{r1r⬘1}{r2r⬘2}i⫽1
冦
兺
36
⫽1
冤
w{r1r⬘1}{r2r⬘2}P
兺
sⱕs⬘ x{ss⬘}
f{ss⬘}(yi)
冥冧
, (9)haplotype frequencies, and the second term to the phe-notype-genotype relationship described by the
geno-where Prob(G{r1r⬘1}{r2r⬘2}) is the population frequency of
typic means and residual variance. The estimates are marker genotypes at the two markers,
then used to update⌸ in the E step, and the process
is repeated until convergence. The values at conver-w{r1r⬘1}{r2r⬘2}⫽
冦
1 if complete categoryis compatible
to marker categoryMr1Mr⬘1Mr2Mr⬘2
0 otherwise
gence are the MLEs.
It is interesting to note that the estimation of allelic frequencies and coefficients of linkage disequilibria can
and the other notation is defined as above. be obtained from the closed-form solutions of haplotype
The estimation of haplotype frequencies is based on frequencies. This will largely improve the computational
the number of a particular haplotype in the population. efficiency of these parameters that can only be
ex-For the complete data, 36 modes of zygote formation pressed as polynomial equations using traditional
proce-via random combinations of paternal haplotypes and
dures (seeLuoandSuhai1999;Luoet al.2000;Wuet
maternal haplotypes are knowna prioriand, therefore,
al.2002).
the number of various marker-QTL haplotypes can be
Disequilibrium mapping of epistatic QTL: We
con-directly counted (see Table 1). But for the incomplete
sider two cases for mapping QTL with epistasis for a data, zygote formation is unobservable and its inference
quantitative trait based on linkage disequilibrium. First, is viewed as a missing data problem. In fact, it is possible
two epistatic QTL are predicted by two independent to calculate the expected number of a particular
marker-pairs of markers. Second, two epistatic QTL are pre-QTL haplotype for a given marker zygote genotype.
dicted by two pairs of genetically associated markers. Although marker-QTL zygote genotypes are also
un-In the first case, the two QTL can be assumed to be known, they can be inferred on the basis of marker
independent from a population genetic standpoint, al-information and phenotype data by implementing the
though they are not so in terms of quantitative genetic EM algorithm. By differentiating Equation 9, we have
effects. In the second case, the two QTL should be associated genetically and interact epistatically to affect
⍀logL(y,M|⍀)⫽r1ⱕ
兺
r1⬘r2ⱕ兺
r⬘2兿
n{r1r⬘1}{r2r⬘2}
i⫽1
兺
36proposed for the above two-marker/one-QTL model is when some regularity conditions are violated. The
per-mutation test approach proposed by Churchill and
extended to estimate both population and quantitative
genetic parameters. Doerge(1994), which does not rely upon the
distribu-tion of the LRQ, may be used to determine the critical
Two basic tasks should be completed before the
im-plementation of the EM algorithm for epistasis map- threshold for claiming the existence of a QTL.
After the existence of the QTL is statistically tested,
ping. The first task is the derivation of an (81 ⫻ 9)
matrix for joint probabilities of four-marker/two-QTL we formulate a second test for its association with two
markers under consideration. The log-likelihood ratio zygote genotypes based on zygote configurations. For
two independent sets of markers and QTL, such proba- for such a test is calculated by
bilities are simply calculated as the Krockner product
LRQ
of the joint probability for each set, so that there are 16
haplotype frequencies (and 14 independent haplotype ⫽ ⫺2 log
冤
L(y,M|p˜k1,pˆk2,pˆs,Dˆ k1k2,Dˆ
s k1⫽Dˆ
s k2⫽Dˆ
s k1k2⫽0,⍀
˜q,˜2)
L(y,M|⍀ˆp,⍀ˆq,ˆ2)
冥
,
frequencies) in this case. But for two dependent sets of
loci, the derivations of such probabilities should con- (14)
which is asymptotically distributed as a2 distribution
sider the formation principle of a six-locus zygote, in
with 3 d.f. which case there are a total of 64 haplotype frequencies,
Other more specific tests for the existence of additive, of which 63 are independent. In the first case, we have
dominant, or epistatic effect and the association of a only digenic and trigenic linkage disequilibria, whereas,
QTL with a particular marker can also be formulated in the second case, we must face digenic, trigenic,
quad-similarly. Generally, these hypotheses can be tested on rigenic, pentagenic, and hexagenic disequilibria (see
the basis of a critical threshold calculated from permuta-Equation 3).
tion tests (Churchill and Doerge 1994). Other
ap-The second task is the modeling of epistatic QTL with
proaches as proposed by Piepho (2001) can also be
additive, dominant, and additive⫻additive, additive⫻
used, which do not require expensive computations.
dominant, dominant⫻additive, and dominant⫻
domi-nant interaction effects (see matrix 4). The estimation of both population (from the first task) and quantitative
MONTE CARLO SIMULATION genetic parameters (from the second task) for epistatic
QTL can be obtained using closed forms. Simulation scenarios: We have performed extensive
simulation studies to investigate the robustness and
per-Hypothesis tests: We can test two major hypotheses
in the following sequence: (1) the existence of QTL formance of our linkage disequilibrium-based mapping
of quantitative traits. A number of simulation scenarios by testing the significance of QTL effects and (2) the
association of significant QTL with markers by testing are performed to compare results under different QTL
models (one-marker/one-QTL vs. multiple-marker/
linkage disequilibrium. The test for the existence of
a significant QTL may not rely upon the association multiple-QTL), different heritability levels, different
sample sizes, and different population genetic parame-between the QTL and known marker(s), but the
ran-dom association of a QTL with markers can be tested ters (equal vs. extreme allele frequencies at a locus).
Marker and QTL genotype data are simulated on the only when the existence of the QTL is statistically tested.
The existence of a QTL can be tested on the basis of basis of joint frequencies for a given sample size, whereas
phenotype data are simulated by summing genotypic two alternative hypotheses:
values and environmental errors, distributed asN(0,2).
H0:{11}⫽ {12} ⫽ {22} The residual variance2is determined under a given
heritability level. The simulated data are analyzed using
H1: at least one equality does not hold. (12)
the genetic model proposed in this article. Each
simula-The log-likelihood-ratio test statistic for the existence tion scenario was repeated 200 times to estimate the
of a QTL is calculated by comparing the likelihood biases and mean square errors of the MLEs.
values under H1(full model) and H0(reduced model), To show the advantage of our multilocus
disequilib-using rium model, we first perform the analysis on the basis of
a one-marker/one-QTL model (MQ), as used in earlier
LRQ⫽ ⫺2 log
冤
L(y,M|⍀˜p,{11}⫽ {12}⫽ {22},
˜2)
L(y,M|⍀˜p,⍀˜q,ˆ2)
冥
, (13) studies (LuoandSuhai1999;Luoet al. 2000;Wuet al.
2002). The results from MQ are compared with those
where the tilde denotes the MLEs of unknown parame- obtained from a two-marker/one-QTL model (MQM)
ters under H0and the other notation is defined as above. and two epistatic QTL models (two separate sets of
two-The LRQis considered to asymptotically follow a2distri- marker/one-QTL models, denoted by MQM-MQM, and
bution with the degrees of freedom equaling the dif- two dependent sets of two-marker/one-QTL models,
ference of the number of unknown parameters to be denoted by MQMMQM). For each QTL model, we
de-estimated under the two hypotheses. However, the ap- sign two different heritabilities (H2⫽0.1 vs.0.4), two
different sample sizes (n⫽500vs.1000), and two
TABLE 2 mapping (MQM5–MQM8; Table 6) over disequilibrium mapping (MQ5–MQ8; Table 5).
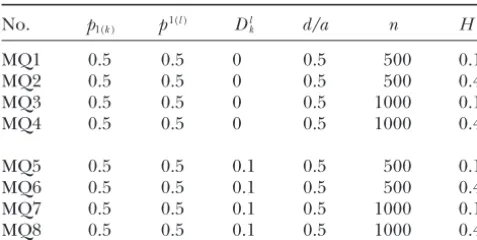
Experimental designs of linkage disequilibrium mapping
We also design a scheme in which two markers are based on the one-marker/one-QTL model (MQ)
associated with each other, both of which are further
No. p1(k) p1(l) Dlk d/a n H2 associated with a putative QTL (MQM9–MQM12).
Al-though this kind of mutual association may not lead to
MQ1 0.5 0.5 0 0.5 500 0.1
a significant improvement for the MLEs of QTL allele
MQ2 0.5 0.5 0 0.5 500 0.4
frequency and residual variance, the estimation for the
MQ3 0.5 0.5 0 0.5 1000 0.1
model mean, the additive and dominant effects of the
MQ4 0.5 0.5 0 0.5 1000 0.4
QTL, and the residual variance is largely benefitted by
MQ5 0.5 0.5 0.1 0.5 500 0.1 a simultaneous use of two associated markers (MQM9–
MQ6 0.5 0.5 0.1 0.5 500 0.4
MQM12; Table 6).
MQ7 0.5 0.5 0.1 0.5 1000 0.1
We examine the effects of different allelic frequencies
MQ8 0.5 0.5 0.1 0.5 1000 0.4
on parameter estimation. The relative frequencies of two alternative alleles reveal the degree of genetic differ-entiation in a population. When QTL alleles are less
ent allele frequencies for a locus. These designs includ- differentiated, as shown by the allele frequencies of
ing the hypothesized parameter values are listed in Ta- 0.3vs.0.7 (MQM13–MQM16), the precision of the MLEs
bles 2–4. is not affected as long as more differentiated markers
Results:Compared to the excellent accuracy and pre- are used (Table 6). We also found that the inclusion of
cision of the MLEs of marker allele frequencies that some less differentiated markers did not affect the MLEs
were obtained directly from the observed data, the accu- of QTL parameters (MQM17–MQM20). However, when
racy and precision of the MLEs of QTL-allele frequency less differentiated markers are used to predict less
differ-and disequilibrium differ-and QTL effects are relatively re- entiated QTL (MQM21–MQM24), the estimation
preci-duced, sometimes seriously. As expected, the MLEs of sion of QTL effects and model parameters is largely
QTL-related parameters under all given QTL models affected, although this may not be true for the MLEs
from MQ to MQMMQM (Table 2–4) display increased of the population genetic parameters of QTL.
accuracy and precision with increased heritabilities and
Additive-dominant-epistasis model:When two QTL
inter-sample sizes, although the extent of the increase may
act epistatically, we develop two different multilocus be different among the models (Table 5–7). Generally, a
disequilibrium schemes to estimate their epistasis. The more significant improvement of parameter estimation,
first scheme (MQM-MQM) permits simultaneous use of especially for QTL allele frequency and disequilibrium,
two independent sets of markers to predict their
corre-was observed whenH2is increased from 0.1 to 0.4 than
sponding QTL, whereas the second scheme (MQMMQM)
whenNis increased from 500 to 1000.
assumes two dependent sets of marker-QTL
relation-Additive-dominant model:Under MQ, we consider two
ships. In general, more tightly linked loci tend to have situations, one with no disequilibrium between the
a greater chance for nonrandom association than less marker and QTL (MQ1–MQ4) and the other with
dis-tightly linked loci do. Thus, the first scheme is roughly equilibrium between the two loci (MQ5–MQ8). The
similar to a situation in which two sets of markers and first situation is similar to the detection of major genes
QTL are from different chromosomes, whereas the sec-because marker information is actually not used. It is
ond scheme is similar to a situation in which two sets found that the MLEs of all QTL-related parameters in
of loci are from the same chromosome. the second situation are improved over those in the first
In the first scheme, all genetic parameters can be situation, especially for the dominant effect of the QTL
estimated with reasonable accuracy and precision (Ta-and model mean (Table 5), suggesting the value of
ble 7), suggesting that our disequilibrium-based map-utilizing marker information in quantitative genetic
re-ping strategy can be used to map epistatic QTL. When search.
two independent marker pairs are used at the same In MQM, we first assume that both markers are not
time, the MLEs of allele frequencies and QTL-associated with a putative QTL (MQM1–MQM4). As
marker disequilibrium are more precise than when only expected, this has no improvement in parameter
estima-one marker pair is used. The root mean-square errors of tion compared to MQ1–MQ4 (Table 6). Second, we
these estimates areⵑ0.122 for QTL allele frequencies,
assume that one of the two markers is associated with
0.033 for QTL-marker digenic disequilibrium, and 0.017 the QTL (MQM5–MQM8), whereas the second marker
for QTL-marker trigenic disequilibrium under MQM-is not. ThMQM-is design MQM-is similar to composite interval
map-MQM1 when two independent marker sets are used ping in which those markers outside the marker interval
(Table 7). Yet, the corresponding values are 0.161, carrying a QTL are used as cofactors to control genome
0.045, and 0.023 under MQM9 when one marker pair background. We did not observe any improvement in
TABLE 3
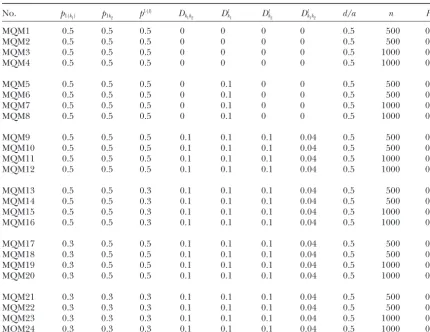
Experimental designs of linkage disequilibrium mapping based on the two-marker/one-QTL model (MQM)
No. p1(k1) p1k2 p
1(l) Dk
1k2 D
l
k1 D
l
k2 D
l
k1k2 d/a n H
2
MQM1 0.5 0.5 0.5 0 0 0 0 0.5 500 0.1
MQM2 0.5 0.5 0.5 0 0 0 0 0.5 500 0.4
MQM3 0.5 0.5 0.5 0 0 0 0 0.5 1000 0.1
MQM4 0.5 0.5 0.5 0 0 0 0 0.5 1000 0.4
MQM5 0.5 0.5 0.5 0 0.1 0 0 0.5 500 0.1
MQM6 0.5 0.5 0.5 0 0.1 0 0 0.5 500 0.4
MQM7 0.5 0.5 0.5 0 0.1 0 0 0.5 1000 0.1
MQM8 0.5 0.5 0.5 0 0.1 0 0 0.5 1000 0.4
MQM9 0.5 0.5 0.5 0.1 0.1 0.1 0.04 0.5 500 0.1
MQM10 0.5 0.5 0.5 0.1 0.1 0.1 0.04 0.5 500 0.4
MQM11 0.5 0.5 0.5 0.1 0.1 0.1 0.04 0.5 1000 0.1
MQM12 0.5 0.5 0.5 0.1 0.1 0.1 0.04 0.5 1000 0.4
MQM13 0.5 0.5 0.3 0.1 0.1 0.1 0.04 0.5 500 0.1
MQM14 0.5 0.5 0.3 0.1 0.1 0.1 0.04 0.5 500 0.4
MQM15 0.5 0.5 0.3 0.1 0.1 0.1 0.04 0.5 1000 0.1
MQM16 0.5 0.5 0.3 0.1 0.1 0.1 0.04 0.5 1000 0.4
MQM17 0.3 0.5 0.5 0.1 0.1 0.1 0.04 0.5 500 0.1
MQM18 0.3 0.5 0.5 0.1 0.1 0.1 0.04 0.5 500 0.4
MQM19 0.3 0.5 0.5 0.1 0.1 0.1 0.04 0.5 1000 0.1
MQM20 0.3 0.5 0.5 0.1 0.1 0.1 0.04 0.5 1000 0.4
MQM21 0.3 0.3 0.3 0.1 0.1 0.1 0.04 0.5 500 0.1
MQM22 0.3 0.3 0.3 0.1 0.1 0.1 0.04 0.5 500 0.4
MQM23 0.3 0.3 0.3 0.1 0.1 0.1 0.04 0.5 1000 0.1
MQM24 0.3 0.3 0.3 0.1 0.1 0.1 0.04 0.5 1000 0.4
precision of QTL additive and dominant effects is re- dominant or dominant⫻additive interaction effect is
similar to the dominant effect, but the dominant ⫻
duced for MQM-MQM (Table 7).
In terms of estimation precision, more precise MLEs dominant interaction effect is poorer than the
domi-nant effect (Table 7). As expected, the MLE of the
of the additive ⫻ additive interaction effect between
two different QTL than those for the additive effect dominant⫻ dominant interaction effect may be quite
imprecise. However, when both sample size and
herita-at a single QTL are obtained, whereas the additive⫻
TABLE 4
Experimental designs of linkage disequilibrium mapping based on two independent sets of two markers and one QTL (MQM-MQM)
No. QTL p1(k1) p1k2 p
1(l) Dk
1k2 D
l k1 D
l k2 D
l
k1k2 d/a i12/a j12/a k12/a l12/a H
2 n
MQM-MQM1 1 0.5 0.5 0.5 0.1 0.1 0.1 0.04 0.5
0.2 ⫺0.2 0.2 ⫺0.2 0.1 500
2 0.5 0.5 0.5 0.1 0.1 0.1 0.04 0.5
MQM-MQM2 1 0.5 0.5 0.5 0.1 0.1 0.1 0.04 0.5
0.2 ⫺0.2 0.2 ⫺0.2 0.4 500
2 0.5 0.5 0.5 0.1 0.1 0.1 0.04 0.5
MQM-MQM3 1 0.5 0.5 0.5 0.1 0.1 0.1 0.04 0.5
0.2 ⫺0.2 0.2 ⫺0.2 0.1 1000
2 0.5 0.5 0.5 0.1 0.1 0.1 0.04 0.5
MQM-MQM4 1 0.5 0.5 0.5 0.1 0.1 0.1 0.04 0.5
0.2 ⫺0.2 0.2 ⫺0.2 0.4 1000
TABLE 5
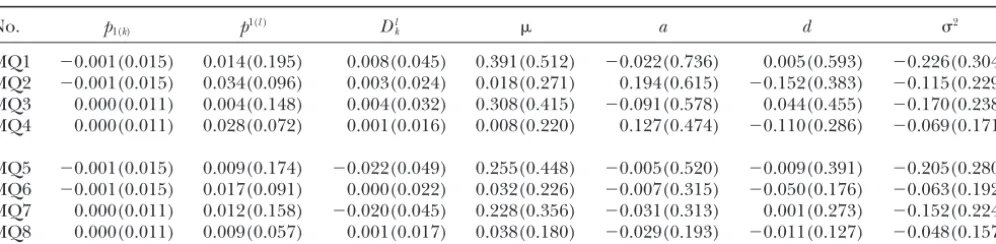
Biases (and squared roots of mean square errors) of genetic parameter estimates based on the one-marker/one-QTL model (MQ)
No. p1(k) p1(l) Dlk a d 2
MQ1 ⫺0.001(0.015) 0.014(0.195) 0.008(0.045) 0.391(0.512) ⫺0.022(0.736) 0.005(0.593) ⫺0.226(0.304)
MQ2 ⫺0.001(0.015) 0.034(0.096) 0.003(0.024) 0.018(0.271) 0.194(0.615) ⫺0.152(0.383) ⫺0.115(0.229)
MQ3 0.000(0.011) 0.004(0.148) 0.004(0.032) 0.308(0.415) ⫺0.091(0.578) 0.044(0.455) ⫺0.170(0.238)
MQ4 0.000(0.011) 0.028(0.072) 0.001(0.016) 0.008(0.220) 0.127(0.474) ⫺0.110(0.286) ⫺0.069(0.171)
MQ5 ⫺0.001(0.015) 0.009(0.174) ⫺0.022(0.049) 0.255(0.448) ⫺0.005(0.520) ⫺0.009(0.391) ⫺0.205(0.280)
MQ6 ⫺0.001(0.015) 0.017(0.091) 0.000(0.022) 0.032(0.226) ⫺0.007(0.315) ⫺0.050(0.176) ⫺0.063(0.192)
MQ7 0.000(0.011) 0.012(0.158) ⫺0.020(0.045) 0.228(0.356) ⫺0.031(0.313) 0.001(0.273) ⫺0.152(0.224)
MQ8 0.000(0.011) 0.009(0.057) 0.001(0.017) 0.038(0.180) ⫺0.029(0.193) ⫺0.011(0.127) ⫺0.048(0.157)
bility are significantly increased, the MLEs of the domi- polymorphisms (SNPs) were genotyped at six candidate
genes for obesity on different chromosomes, which are
nant⫻dominant effect can be close to those of other
parameters (Table 7). ADRB1,ADRB2, ADRB3, ADRA1A, the GSprotein␣
-sub-unit (GNAS1), and the G protein 3 subunit (GNB3;
In the second scheme, the precision of the MLEs of
all parameters is reduced, compared to the first scheme, J. A.Johnson,unpublished results). TheADRB1 gene
contains two common nonsynonymous polymorphisms because of the inclusion of many unknowns. We
per-form an additional simulation underH2⫽ 0.4 for the at codon 49 (Ser→Gly) and codon 389 (Arg→Gly).
At theADRB2 gene, there are three polymorphism sites
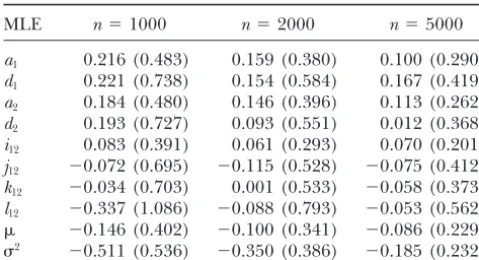
second scheme by increasing the sample size from 1000
to 2000 to 5000, keeping the other parameters un- at codon 19 of the2AR 5⬘leader cistron (Cys→Arg),
codon 16 (Gly→Arg), and codon 27 (Gln→Gly). Two
changed. The precision of the MLEs of population
ge-netic parameters remained approximately constant with sites at codon 131 and codon 371 were genotyped for
theGNAS1 gene.
increased sample sizes (data not given), but the MLEs
of gene effects including the additive, dominant, and Our newly developed statistical method is used to
perform the association studies between SNPs and body epistatic effects displayed significantly improved
preci-sion when the sample size is increased (Table 8). When heights in two different populations. According to
Mackay (2001), the gene for a quantitative trait
de-N⫽2000, the estimation of the additive and additive⫻
additive interaction effects achieves acceptable preci- tected by a SNP is called a quantitative trait nucleotide
sion under the complex MQMMQM model. The estima- (QTN). We found different QTN that affect human
tion of the other effects is certainly acceptable when a body heights between the black and white populations
sample size is 5000 (Table 8). (Table 9). Two SNPs, Gly16Arg andCys19Arg, at gene
ADRB2 can identify significant QTN in the black
popula-tion, although the significance of the QTL detected by
A CASE STUDY FROM HUMANS the second SNP is marginal (Table 9). In fact, the QTL
detected by these two SNPs may be identical for two A case study from a human genome project for
map-reasons. First, the sum of the allele frequencies of the ping obesity genes is used to validate our method. The
QTN identified is one, implying that variants detected subjects sampled consisted of 643 women from the
Na-by the two SNPs within the same gene are actually two tional Heart, Lung, and Blood Institute-funded
Wom-alternative alleles. This Wom-alternative relationship is con-an’s Ischemic Syndrome Evaluation (WISE) study ( J. A.
firmed by different signs of disequilibrium coefficients.
Johnson,unpublished results). Women included in the
Second, the QTN displays a similar genetic effect on
WISE study were⬎18 years old and consist of 105 blacks
body height, with different directions for the additive and 538 whites, each measured for body height and
effect as expected. This black-specific height QTN can other traits. However, as an example, only body height is
also be identified by combining one of these two SNPs analyzed here. All study participants provided informed
with SNPC825Tat geneGNB3.
written consent prior to participation in the study. WISE
In the white population, a significant QTN was de-study protocols were approved by the Institutional
Re-tected by SNPC835Tat geneGNB3 (Table 9). As shown
view Boards of the participants’ institutions.
by allele frequencies, linkage disequilibria, and genetic Genotypes for the women studied were determined
effects, we conjecture that the same QTN has been de-using Orchid BioScience’s proprietary primer extension
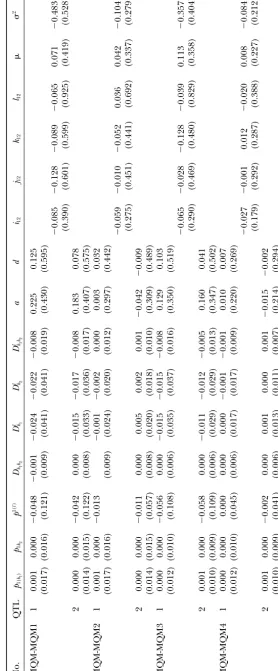
TABLE 6 Biases (and squared roots o f m ean square errors) of genetic parameter estimates based on the two-marker/one-QTL model (MQM) No. p1( k1 )
p1k
2 p 1( l ) Dk 1 k2 D
l k1
D
l k2
D
l kk1
TABLE 7 Biases (and squared roots o f mean square errors) of genetic parameter estimates based on two independent sets of two m arkers and one QTL (MQM-MQM) No. QTL p1( k1 )
p1k
2 p 1( l ) Dk 1 k2 D
l k1
D
l k2
D
l kk12