78:6–4 (2016) 1–6 | www.jurnalteknologi.utm.my | eISSN 2180–3722 |
Jurnal
Teknologi
Full Paper
S
OME
I
NTRIGUING
H
IGH
-T
HROUGHPUT
DNA
S
EQUENCE
V
ARIANTS
P
REDICTION
O
VER
P
ROTEIN
F
UNCTIONALITY
Atabak Kheirkhah, Salwani Mohd Daud
*, Noor Azurati Ahmad @
Salleh, Suriani Mohd Sam, Hafiza Abas, Sya Azmeela Shariff,
Yusnaidi Md Yusof
Advanced Informatics School (AIS), Universiti Teknologi Malaysia,
54100 UTM Kuala Lumpur, Malaysia
Article history Received 10 July 2015 Received in revised form
2 December 2015 Accepted 16 January 2016
*Corresponding author
[email protected]
Graphical abstract
Abstract
This paper intends to review computational methods and high throughput automated tools for precisely prediction various functionalities of uncharacterized proteins based on their desired DNA sequence information alone. Then proposes a hybrid weighted network and Genetic Algorithm to improve prediction purpose. The main advantage of the method is the protein function and DNA sequence prediction can be computed precisely using best fitness parent in genetic algorithm. With the accomplishment of human genome sequencing, the number of sequence-known proteins has increased exponentially and the pace is much slower in determining their biological attributes. The gap between DNA sequence variants and their functionalities has become increasingly large. However, detection of sequences based on protein data bank has become benchmark for many researchers. As amount of DNA sequence data continues to increase, the fundamental problem stay at the front of genome analysis. In the course of developing these methods, the following matters were often needed to consider: benchmark dataset construction, gene sequence prediction, operating algorithm, anticipated accuracy, gene recommender and functional integrations. In this review, we are to discuss each of them, with a different focus on operational algorithms and how to increase the accuracy of DNA sequence variants prediction.
Keywords: DNA Sequence variants, protein interactions, protein functional integration
© 2016 Penerbit UTM Press. All rights reserved
1.0 INTRODUCTION
The main part of any gene prediction system is determining what types of interactions are most relevant to a given query list[1]. Hence, the interactions among the proteins are necessity for different biological functions in living cells. Establishing knowledge about these interactions offers a basis to lead protein interaction networks and improves our considerate of the general principles of the gene and cancer functionality.
might depend on biological context. In comparison the storage of protein-protein associations in databases is less straightforward than genomic sequence data and other types as so on [12].
For a wide range of model organisms, genome-scale networks exist in the physical interactions among proteins. As organize large networks of synthetic genetic interactions [13], miRNA-mRNA interactions (both computationally predicted [14], measured [15]) protein-DNA [16], and protein-RNA [17] interactions. The functional interactions between proteins may be construed from interactions between their orthologous gene in other species [18, 19], also their sequence similarity, pattern of co-expression [19, 20], shared mutant phenotypes, protein domain structure, or shared subcellular localizations. Each of indicated interaction networks on its own presents a distinct situation, but probably piercing and unfair [1].
With the fast growth of protein sequences generated in the post-genomic age, researchers are trying to know their attributes because they are closely correlated with the structures and functions of the proteins as well as their roles in biological processes. Hence are very important to both basic research and drug target development [21]. For instance, given an uncharacterized protein sequence, what is its folding rate? Which structural class and quaternary structural attribute does it belong to? Is it a protease? If it is, to which protease type does it belong? Which part of the protein serves as its signal sequence? How can predict the healthy genome? And so forth. Although the answers to these questions can be determined by leading various biochemical experiments, it is both costly and time-consuming by relying on experimental approaches alone [22]. Consequently, the gap between the number of latest discovered protein sequences, the knowledge of their attributes and the precise prediction of their computational method is continuing to expand. To bridge such a gap and obtain these kinds of information in a timely manner,
researchers are challenged to develop
computational methods for predicting various attributes of proteins based on their sequence information alone.
Most of the computational methods require not only sequence information but also auxiliary data i.e. localization data, expression data, structural data. Indeed, many records in this regard have been established in last two decades in hope to cover such a gap. In course of developing these methods, the following matters were often needed to consider:
• gene prediction, operating algorithm,
• gene recommenders,
• functional integration network
The main contribution of this review is to investigate the above procedures, with a special focus on current systems algorithms to compare the protein prediction functionalities.
2.0 GENE PREDICTION
Prediction is providing an enquiry list of DNA sequence that shows some feature of their functions. The systems predicting gene to anticipate other genes that are probable to be co-functional with the genes that existed inside the query list [1]. These predictions are prepared by discovering other DNA sequence variant highly connected to the query list in a composite function integration network (or simple composite network), which is concluded using data from many studies. Classically, the composite network is generated by combining several other interaction networks derived from one of the following sources:
(i) genome-scale studies of gene and protein interactions
(ii) curation of several small-scale studies of gene and protein interactions
(iii) automated procedures that infer functional interactions among gene pairs
The desired approach would be protein modelling which use a frame-based demonstration. Interactions between the amino acids and proteins are conveyed by utilizing the production rules and an underlying truth maintenance system. As proposed in many studies a graphical interface is provided access to all elements of the simulation, comprised of object representation and explanation graphs [5, 23, 24].
To get the desired outcomes, the sequence similarity search based approach is very important. For example Basic Local Alignment Search Tool (BLAST), which is a rapid sequence comparison tool utilized a heuristic approach to construct alignments by optimizing a measure of local similarity. BLAST compares DNA sequences much faster than dynamic programming methods such as
Smith-Waterman and Needleman-Wunsch [25]
approaches. Moreover, there are many tools following BLAST’s approach as a standard such as Argot2 [26].
3.0 PREDICTION ALGORITHMS
There are several prediction algorithms were introduced, such as neural network algorithm[27, 28], support vector machine (SVM) [29, 30] and K-nearest Neighbor algorithm [28, 29, 31].
As well, Rotamer libraries are containing of information on side-chain prediction methods based on backbone-dependent approach. In side-chain prediction method, which is based on protein sequence and given backbone coordinates, utilizes a search strategy and defined energy function to predict the nearest solution.
3.1 Neural Network Algorithm
Neural network is a very useful algorithm for biological data mining due to their adaptive nature with a pervasive utilized area [32-34]. The approach of neural network is based on learning from examples and preparing desired technique suitable for issues such as secondary protein structure prediction and TM domain prediction. Therefore, performance of neural network is depended on availability of high quality training data. According to previous experience [32] there are two kinds of network topologies that is feed-forward and feed-back, which currently, feed-forward neural network is the most utilized technique.
Neural network is a non-linear mathematical approach, which is a popular algorithm in pattern recognition and computes the data using a connectionist approach. It is one of the first techniques used for protein recognition [33, 35].
Indeed, in feature-based approaches, information about function is assumed to be predictable via a range of features of proteins. The features are comprised of secondary structure, port-translational modifications and general properties of amino acid composition [36].
In additional, the training process of a neural network is to amend the network weights according to a learning algorithm until the failure of network gets its minimum. The trained network (comprised of important information of weights) has the function of identifying the domain structure class [27].
3.2 Support Vector Machine Algorithm
Support Vector Machine (SVM) has been extensively utilized in solving prediction problems in bioinformatics, which represented in practice of a binary classification, i.e. gene identification, gene detection and protein-protein interaction prediction [32, 37].
As stated earlier, SVMs are kinds of learning machines based on statistical learning theory. Briefly, applying SVMs in pattern classification is according to the following steps:
(i) Mapping the input vectors into one feature space, either linear or nonlinearly.
(ii) Seeking an optimized linear division within the feature space from the first step.
Indeed, the SVM training process always pursues an overall optimized solution and avoids over-fitting, therefore it is able to be employed with a large number of features.
Kazemian et al. [32] proposed a new methodology using SVM based on statistical encoding technique to predict trans-membrane protein such as helices. In comparison with the number of original training data pairs, the total number of support vector is small. They have combined SVM with another AI algorithm such as Genetic Algorithm to improve the accuracy of prediction till 87% [32].
Chuanxin Zou et al. [38] demonstrate an implementation of SVM based on publically available LIBSVM package version 3.11 [39] and according to grid search method to deliver high accuracy, the tunable parameters are optimized.
3.3 Proposed Algorithm for Enhancing the Accuracy
Table 1 is a comparison for some existing approaches. However, if a structure is available for the target protein, the algorithm should check whether it reveals high similarity to other DNA sequence which already identified structures in complex of proteins. Additionally, the DNA sequence may be utilized to model the protein structure computationally, in particular using the comparative view. For this, the study proposes the hybrid of weighted network and Genetic Algorithm (GA) to achieve a precise undesired mutation detector.
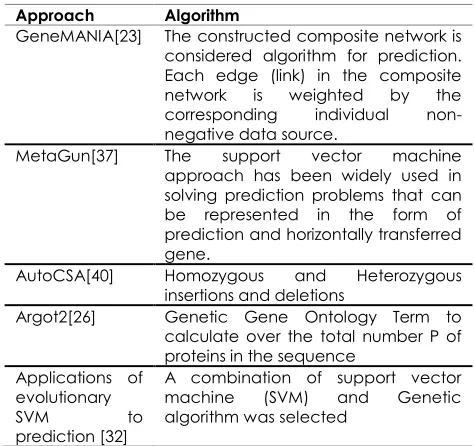
Table 1 A comparison for some existing approaches
Approach Algorithm
GeneMANIA[23] The constructed composite network is considered algorithm for prediction. Each edge (link) in the composite network is weighted by the corresponding individual non-negative data source.
MetaGun[37] The support vector machine approach has been widely used in solving prediction problems that can be represented in the form of prediction and horizontally transferred gene.
AutoCSA[40] Homozygous and Heterozygous insertions and deletions
Argot2[26] Genetic Gene Ontology Term to calculate over the total number P of proteins in the sequence
Applications of evolutionary SVM to prediction [32]
A combination of support vector machine (SVM) and Genetic algorithm was selected
GA begins with a set of chromosomes as candidate solutions for population. New populations are created from old ones in hope to achieve a better population. The solutions which are chosen to form new offspring are opted based on their fitness. Current classical studies are utilizing just amino acid sequence and some model of the interaction between amino acids to determine structures[25, 41]. Therefore focusing from two points of view is outstanding: first is development of methods to search for the functional conformation and second development of more reliable ways of discriminating between right and wrong structures.
to assess fitness and apply GA on precise prediction. The comprehensive benchmark dataset arranged based on DNA sequence similar functionality would play a prominent role in GA process to enhance the accuracy.
The outcome of the model can be used as a query for structure-based methods. Nevertheless, the theoretical view usually contains various defects and inaccuracies. Indeed, a theoretical view should be first evaluated by Quality Assessment Models through the Model Quality Assessment Programs.
3.4 A Hybrid View on Weighted Network and Genetic Algorithm
As stated earlier a proposed hybrid approach of weighted network and genetic algorithm may improve the current situation of gene predictions. It looks promising by focusing on population and find the fitness gene the prediction of protein interaction will be affected by time and cost matters. Genetic Algorithm as a robust algorithm has been proven to efficiently search in complex solution spaces. Practicing DNA sequence variants with GA reveals flexibility of search algorithm. Genetic Algorithm generates new feasible collections and population to computes every possible sorting of sequenced and genes. Depend on the fitness parent for each crossover and mutation the result will be more measurable for protein functionality prediction. Figure 1 shows the weighted networks for two sample genes and other expression genes.
Figure 1 Weighted networks for two sample genes and other expression genes
Assume there is a set of genes S = [s1, s2… sn], illustrated in a weighted network based on expressions. Figure 2 shows a graphical view of genes interactions between two genes with others.
Every reproduction from these two networks illustrates the survivals from the fitness selection process during the genetic algorithm flow. Figure 3 shows two sequences considered for cross over. If during the selection process genes comes from the fitness parent, therefore outcome reach to a survival scope, even utilize for next patent selection and the rest of genes with low fitness values would be excluded. Consequently, by a focus on weighted network and search for fitness nodes the genetic algorithm flow is improved to concentrate on fitness
nodes and eliminate the undesired nodes during the selection stage.
S1 S2 S3 S4 S5 S6 S7 … Sn
S1 S2 S3 S4 S5 S6 S7 … Sn
Figure 2 An example of extracted sequences for cross over
The significant section of process is proceeding to find the fitness values based on existing weights between the nodes which help to select the best parents. Moreover by a recursive view on GA after each step the population reach to a precise prediction. Therefore, depend on the fitness population from every step the follow process obtain a better protein functionality prediction based on the similar sequences.
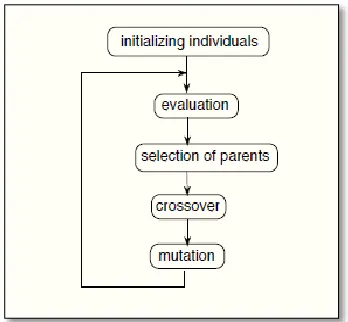
Figure 3 shows an overview on GA process with an included loop. Proposed hybrid algorithm has a main establishment on evaluation to generate weighted network and after that selecting the fitness parent effect on every crossover and mutation.
Figure 3 Over view on genetic algorithm’s flow
Although some approach such as Routing Wheel have a selection based on largest weights but have a fitness population for DNA sequence variants is more affected from similarities related to nodes weights. The summarized pseudo code for prediction of protein functionality based on genetic algorithm will be as follows:
initial_population () Evaluate () While (population)
generate_weightedNetwork (new population) select_FitnessParents (population)
Recombine () // proceed Crossover Mutation ()
Accordingly, for every execution of loop the process products a new population and depend on the fitness parent the result will be different. It is significant that crossover needs to be execute for genes within the sequence, and if the length of effected sequence become shorter the new population converge to the more fitness parents for prediction.
By replacing the genes in new network and recombine the results, complexity of algorithm reach to a better performance by O (nlog n). In addition, number of nodes after each time execution of loop is important.
4.0 CONCLUSIONS AND PERSPECTIVES
According to huge amount of protein sequence and interactions, discovering post-genomic era for disease and drug research is occupied the vital zone of biological development studies. Many studies around the word attempt to utilize the computational methods, and mathematical analysis to handle the biology and medicine concepts, such as graphical analysis [18, 28, 35], model of gene regulatory networks [42], investigate functional solutions [1, 32]. Therefore, it’s highly required to investigate, analyze and develop an automated method to introduce a fast and precise predictor for DNA sequence variants. In this review the fundamental section efforts to focus on exists prediction algorithms in hopes that the points enhanced here may help anticipating the further development of more powerful predictors. It is proposed that a hybrid circumstances of those algorithm such as Genetic algorithm and weighted network aid to predict most functionalities faster and more powerful. Moreover an approach on bio inspired algorithms such as genetic algorithm aid to simulate a manipulatable environment to predict. However, the matter of this approach is investigation of using genetic algorithm for protein functionality according to DNA sequence variants more than focus on selection process which will come in next research. How can achieve to a fast and precise selection by eliminate the redundant node after each step.
Acknowledgement
We would like to express our gratitude to Ministry of Higher Education (MOHE Malaysia) for providing
financial support (research grant
R.K130000.7838.4F357) in conducting our study. Last but not least, we would also like to express our appreciation to Universiti Teknologi Malaysia (UTM) and specifically Advanced Informatics School (AIS) for realizing and supporting this research work.
References
[1] Mostafavi S. and Morris Q. 2012. Combining Many Interaction Networks To Predict Gene Function And Analyze Gene Lists. Proteomics. 12: 1687-1696.
[2] Pattin, K. A. and Moore, J. H. 2009. Role For Protein–Protein Interaction Databases In Human Genetics. Expert Review Of Proteomics. 6: 647-659.
[3] Colinge, J., Rix, U., Bennett, K. L., and Superti‐Furga, G.
2012. Systems Biology Analysis Of Protein‐Drug Interactions. PROTEOMICS-Clinical Applications. 6: 102-116.
[4] Pujol, A., Mosca, R., Farrés, J., and Aloy, P. 2010. Unveiling The Role Of Network And Systems Biology In Drug Discovery. Trends In Pharmacological Science. 31: 115-123.
[5] Brutlag, D. L., Galper, A. R., and Millis, D. H. 1991. Knowledge-based Simulation Of DNA Metabolism: Prediction Of Enzyme Action. Computer Applications In The Biosciences: CABIOS. 7: 9-19.
[6] Kreeger, P. K. and Lauffenburger, D. A. 2010. Cancer Systems Biology: A Network Modeling Perspective. Carcinogenesis. 31: 2-8.
[7] Syed, A. S., D’Antonio, M., and Ciccarelli, F. D. 2009. Network Of Cancer Genes: A Web Resource To Analyze Duplicability, Orthology And Network Properties Of Cancer Genes. Nucleic Acids Research. 957.
[8] Janga, S., Díaz-Mejía, J. J., and Moreno-Hagelsieb, G.
2011. Network-based function Prediction And
Interactomics: The Case For Metabolic Enzymes. Metabolic Engineering. 13: 1-10.
[9] Orth, J. D. and. Palsson, B. Ø. 2010. Systematizing The
Generation Of Missing Metabolic Knowledge.
Biotechnology And Bioengineering. 107: 403-412.
[10] Tsoka, S. 2007. Computational Methodologies For
Genome Evolution And Functional Association.
Computers & Chemical Engineering. 31: 943-949.
[11] Wang, P. I. and Marcotte, E. M. 2010. It's The Machine That Matters: Predicting Gene Function And Phenotype From Protein Networks. Journal Of Proteomics. 73: 2277-2289. [12] Szklarczyk, D., Franceschini, A., Kuhn, M., Simonovic, M.,
Roth, A., Minguez, P., et al. 2011. The STRING database In 2011: Functional Interaction Networks Of Proteins, Globally Integrated And Scored. Nucleic Acids Research. 39: D561-D568.
[13] Costanzo, M., Baryshnikova, A., Bellay, J., Kim, Y., Spear, E. D., Sevier, C. S., et al. 2010. The Genetic Landscape Of A Cell Science. 327: 425-431.
[14] Betel, D., Wilson, M., Gabow, A., Marks, D. S., and Sander, C. 2008. The microRNA. Org Resource: Targets And Expression. Nucleic Acids Research. 36: D149-D153. [15] Hsu, R.-J. and Tsai, H.-J. 2011. Performing the Labeled
microRNA Pull-down (LAMP) assay System: An
Experimental Approach For High-Throughput Identification Of Microrna-Target mRNAs. Therapeutic Oligonucleotides. ed: Springer. 241-247.
[16] Birney, E., Stamatoyannopoulos J. A., Dutta A., Guigó R., Gingeras T. R., Margulies E. H., et al. 2007. Identification And Analysis Of Functional Elements In 1% Of The Human Genome By The ENCODE Pilot Project. Nature. 447: 799-816.
[17] Hafner, M., Landthaler, M., Burger, L., Khorshid, M., Hausser J., Berninger P., et al. 2010. Transcriptome-wide Identification Of RNA-Binding Protein And Microrna Target Sites By PAR-CLIP. Cell. 141: 129-141.
[18] Jerlström-Hultqvist, J., Franzén, O., Ankarklev, J., Xu, F., Nohýnková, E., Andersson, J. O., et al. 2010. Genome Analysis And Comparative Genomics Of A Giardia Intestinalis Assemblage E Isolate. BMC Genomic. 11: 543. [19] Pellegrini, M., Marcotte, E. M., Thompson, M. J., Eisenberg,
[20] Brown, M. P., Grundy, W. N., Lin, D., Cristianini, N., Sugnet C. W., Furey T. S, et al. 2000. Knowledge-based Analysis Of Microarray Gene Expression Data By Using Support Vector Machines. Proceedings of the National Academy of Sciences. 97: 262-267.
[21] Chou K.-C. 2011. Some Remarks On Protein Attribute Prediction And Pseudo Amino Acid Composition. Journal Of Theoretical Biology. 273: 236-247
[22] De Wit, M., Junginger, M., Lensink, S., Londo, M., and Faaij, A. 2010. Competition Between Biofuels: Modeling Technological Learning And Cost Reductions Over Time. Biomass And Bioenergy. 34: 203-217,
[23] Warde-Farley, D., Donaldson, S. L., Comes, O., Zuberi, K., Badrawi, R., Chao, P., et al. 2010. The GeneMANIA Prediction Server: Biological Network Integration For Gene Prioritization And Predicting Gene Function. Nucleic Acids Research. 38: W214-W220.
[24] Rhead, B., Karolchik, D., Kuhn, R. M., Hinrichs, A. S., Zweig, A. S., Fujita, P. A. et al. 2009. The UCSC Genome Browser Database: Update 2010. Nucleic Acids Research. 939. [25] Needleman, S. B. and Wunsch, C. D.1970. A General
Method Applicable To The Search For Similarities In The Amino Acid Sequence Of Two Proteins. Journal Of Molecular Biology. 48: 443-453,
[26] Falda, M., Toppo, S., Pescarolo, A., Lavezzo, E., Di Camillo, B., Facchinetti, A. et al. 2012. Argot2: A Large Scale Function Prediction Tool Relying On Semantic Similarity Of Weighted Gene Ontology Terms. BMC Bioinformatics.13: S14.
[27] Cai, Y.-D. and Chou, K.-C. 2000. Using Neural Networks For Prediction Of Subcellular Location Of Prokaryotic And Eukaryotic Proteins. Molecular Cell Biology Research Communications. 4: 172-173.
[28] Dehouck, Y., Grosfils, A., Folch, B., Gilis, D., Bogaerts, P., and Rooman M. 2009. Fast And Accurate Predictions Of Protein Stability Changes Upon Mutations Using Statistical
Potentials And Neural Networks: PoPMuSiC-2.0.
Bioinformatic. 25: 2537-2543.
[29] Cai, Y.-D., Ricardo, P.-W., Jen, C.-H., and Chou, K.-C. 2004. Application Of SVM To Predict Membrane Protein Types. Journal of Theoretical Biolog. 226: 373-376.
[30] Kumar, M., Gromiha, M. M., and Raghava, G. P. 2011. SVM Based Prediction Of RNA‐Binding Proteins Using Binding
Residues And Evolutionary Information. Journal of Molecular Recognition. 24: 303-313.
[31] Tegge, A. N., Wang, Z., Eickholt, J., and Cheng, J. 2009. NNcon: Improved Protein Contact Map Prediction Using 2D-Recursive Neural Networks. Nucleic Acids Research. 37: W515-W518,
[32] Kazemian, H. B., White, K., and Palmer-Brown, D. 2013. Applications Of Evolutionary SVM To Prediction Of
Membrane Alpha-Helices. Expert Systems with
Applications. 40: 3412-3420.
[33] Seguritan, V., Alves, Jr N., Arnoult, M., Raymond, A., Lorimer, D., Burgin, Jr A. B., et al. 2012. Artificial Neural Networks Trained To Detect Viral And Phage Structural Proteins.
[34] Bose, S. K., Kazemian, H., Browne, A., and White, K. 2006. Presenting A Novel Neural Network Architecture For Membrane Protein Prediction. in Intelligent Engineering Systems, 2006. INES'06. Proceedings. International Conference.135-138.
[35] Volpato, V., Adelfio, A., and Pollastri, G. 2013. Accurate Prediction Of Protein Enzymatic Class By N-To-1 Neural Networks. BMC bioinformatics. 14: S11.
[36] Whisstock, J. C. and Lesk, A. M. 2003. Prediction Of Protein Function From Protein Sequence And Structure. Quarterly Reviews Of Biophysics. 36: 307-340.
[37] Liu, Y., Guo, J., Hu, G., and Zhu, H. 2013. Gene Prediction In Metagenomic Fragments Based On The SVM Algorithm. BMC Bioinformatics. 14: S12.
[38] Zou, C., Gong, J., and Li, H. 2013. An Improved Sequence Based Prediction Protocol For DNA-Binding Proteins Using
SVM And Comprehensive Feature Analysis. BMC
Bioinformatics. 14: 90.
[39] Chang, C.-C.and Lin, C.-J. 2011. LIBSVM: A Library For Support Vector Machines. ACM Transactions On Intelligent Systems And Technology (TIST). 2: 27.
[40] Dicks, E., Teague, J. W., Stephens, P., Raine, K., Yates, A., Mattocks C., et al. 2007. AutoCSA, An Algorithm For High Throughput DNA Sequence Variant Detection In Cancer Genomes. Bioinformatics. 23: 1689-1691.
[41] Qi, J.-P., Shao, S.-H., Li, D.-D., and Zhou, G.-P. 2007. A Dynamic Model For The P53 Stress Response Networks Under Ion Radiation. Amino Acids. 33: 75-83.