ABSTRACT
BASNET, LOCHAN. Uncertainty and Sensitivity Analysis of Dynamic Biological Systems under Data Sparseness. (Under the direction of Joel J. Ducoste)
Systems biology integrates experimental observations with mathematical models to infer
the behavior of biological organisms at the genetic and metabolic levels. Ordinary
Differential Equation (ODE) based dynamic models are frequently used to describe these
biological behaviors under dynamic and steady state conditions. ODE-‐based models are
flexible in structure but may involve a large number of parameters. Under data sparseness
from the lack of experimental replicates under transient conditions, the parameters that are
estimated have uncertainties associated with them. Parameter uncertainties get
propagated through the model into the outputs resulting in misleading predictions and
inferences. A comprehensive understanding of these uncertainties can, however, prevent
such misleading deductions as well as provide valuable information about the behavior of
the biological systems. Given the scope of dynamic models in biological studies, such
comprehensive uncertainty studies for these models are lacking.
In this research, our aim was to perform a comprehensive uncertainty study of ODE-‐based
dynamic models under data sparseness. Through such study, our goal was to quantify the
uncertainty in the model parameters and outputs, assess the identifiability of these
output uncertainties under data sparseness. We studied two ODE-‐based dynamic models:
the yeast synthetic network model in Saccharomyces cerevisiae and a preliminary iron deficiency response model in Arabidopsis thaliana. In this research, an assessment of the accuracy of these methods was provided to deduce the biological information for both
systems. As part of this assessment, we demonstrated the applicability of the Morris
Screening method for sensitivity analysis of dynamic models. Through the bootstrap
method of uncertainty analysis, we calculated confidence intervals that encapsulated true
parameter values as well as the true expression of the genes in the yeast synthetic network
model. Assessment of practical identifiability using profile likelihood showed that the data
sparseness was high for both system models. Finally, the Morris Screening method was able
to identify the important and the unimportant parameters based on their contribution to
the output uncertainties for both of the models. Based on the results from Morris
Screening, we were able to explain the regulatory effects of SWI5 and ASH1 genes on CBF1
gene in the yeast synthetic network model. For the iron transcription factors in the
Arabidopsis thaliana gene regulatory network, we identified a clear influence of iron effect in the gene expression for COL4 and of constitutive transcription and degradation in the
gene expression of bHLH104, however, similar deductions could not be made for the
remaining transcription factors in the network. Sensitivity results identified the least
important parameters in the yeast synthetic network model that could be screened out for
the purpose of addressing data sparseness in any future analyses.
Ó Copyright 2017 Lochan Basnet
Uncertainty and Sensitivity Analysis of Dynamic Biological Systems under Data Sparseness
by Lochan Basnet
A thesis submitted to the Graduate Faculty of North Carolina State University
in partial fulfillment of the requirements for the Degree of
Master of Science
Civil Engineering
Raleigh, North Carolina
2017
APPROVED BY:
_________________________ _________________________
Cranos M. Williams, PhD Ranji Ranjithan, PhD
_________________________ Joel J. Ducoste, PhD
DEDICATION
To my sister Ritu Basnet,
Thank you for the support, love and guidance in every walk of my life.
To my mother and father,
Thank you for your love, care and all the opportunities you have provided that I feel blessed of.
BIOGRAPHY
Lochan Basnet was born in Pokhara, Nepal. He completed his undergraduate studies
as a Civil Engineer in year 2013 from Nepal. In the fall of 2015, he joined North Carolina
State University and began his pursuit of Master of Science in Civil Engineering under the
guidance of Dr. Ducoste.
ACKNOWLEDGEMENTS
I would first like to thank my advisor Dr. Joel J. Ducoste for providing me the opportunity to
be a part of such wonderful research. I am grateful for his continuous support,
encouragement, guidance, and valuable advice without which this thesis would not have
been possible. You are a great teacher and a wonderful person.
I also want to thank my thesis committee member and the principal investigator for the
research Dr. Cranos M. Williams for the critical analysis of my work and the valuable insights
that has helped me throughout my study. I am very grateful to the entire INSPIRE group for
providing me the data and the model used in this study.
I am thankful to Dr. Ranji Ranjithan for taking time to serve in my thesis committee. I am
also thankful to all the faculty and staff of the Department of Civil Engineering. Many thanks
to Renee Howard for all the timely information that helped me remain in right track
throughout the pursuit of my Master’s degree.
I would like to thank the National Science Foundation for the financial support.
TABLE OF CONTENTS
LIST OF TABLES ... vii
LIST OF FIGURES ... viii
SUPPLEMENTAL TABLES ... ix
SUPPLEMENTAL FIGURES ... x
CHAPTER 1 ... 1
Introduction ... 1
1.1 Objectives ... 3
CHAPTER 2 ... 5
Background ... 5
2.1 Dynamic system models and uncertainty ... 5
2.2 Uncertainty Analysis ... 5
2.2.1 Non-‐sampling based Uncertainty Analysis ... 7
2.2.2 Sampling based Uncertainty Analysis ... 8
2.3 Sensitivity Analysis ... 10
2.3.1 Local Sensitivity Analysis ... 11
2.3.2 Global Sensitivity Analysis ... 12
2.4 Preliminary Iron deficiency response model in Arabidopsis thaliana ... 14
CHAPTER 3 ... 16
Methods ... 16
3.1 Model ... 16
3.1.1 Yeast Synthetic Network Model in Saccharomyces cerevisiae ... 16
3.1.2 Preliminary Iron Deficiency Response Model in Arabidopsis thaliana ... 19
3.2 Bootstrap Sampling for Data Generation ... 19
3.3 Parameter Estimation ... 21
3.4 Uncertainty Analysis ... 21
3.5 Identifiability Analysis ... 22
3.6 Sensitivity Analysis ... 23
3.6.1 Extension to Morris Method ... 24
CHAPTER 4 ... 26
4.1 Introduction ... 27
4.2 Background ... 30
4.2.1 Dynamic system models and uncertainty ... 30
4.2.2 Uncertainty Analysis ... 30
4.2.3 Sensitivity Analysis ... 32
4.2.4 Iron deficiency response model in Arabidopsis thaliana ... 33
4.3 Methods ... 35
4.3.1 Model ... 35
4.3.2 Bootstrap Sampling for Data Generation ... 37
4.3.3 Parameter Estimation ... 39
4.3.4 Uncertainty Analysis ... 39
4.3.5 Identifiability Analysis ... 39
4.3.6 Morris Screening for Sensitivity Analysis ... 40
4.4 Results ... 42
4.4.1 Yeast Synthetic Model in Saccharomyces cerevisiae ... 42
4.4.2 Iron Deficiency Model in Arabidopsis thaliana ... 48
4.5 Discussion ... 55
4.6 Conclusion ... 58
CHAPTER 5 ... 59
Conclusion and Future Work ... 59
BIBLIOGRAPHY ... 62
APPENDIX ... 74
LIST OF TABLES
Table 2.1: Comparison of frequently used UA techniques ... 7
Table 2.3: Comparison of commonly used SA methods ... 11
Table 4.1: Parameters ranked by Savage scores under Fe-‐ condition ... 55
LIST OF FIGURES
Figure 1: Iron Deficiency Gene Regulatory Network in Arabidopsis thaliana: ... 15
Figure 2: Yeast Synthetic Network in Saccharomyces cerevisiae: ... 18
Figure 4.1: Iron Deficiency Gene Regulatory Network in Arabidopsis thaliana: ... 34
Figure 4.2: Yeast Synthetic Network in Saccharomyces cerevisiae: ... 36
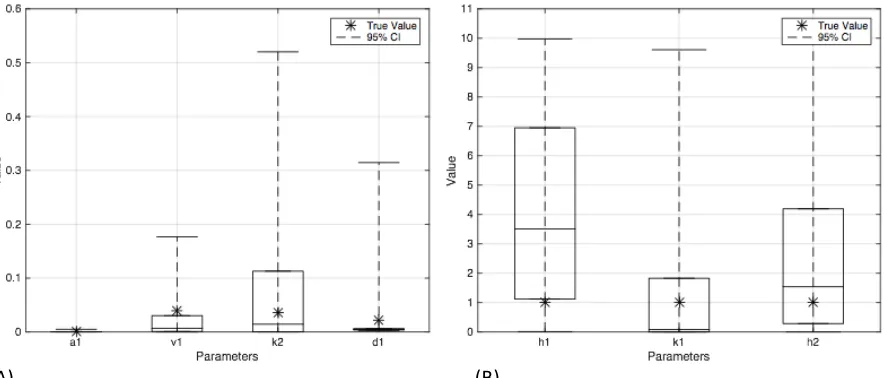
Figure 4.3 Boxplots of few parameters in Yeast synthetic model ... 43
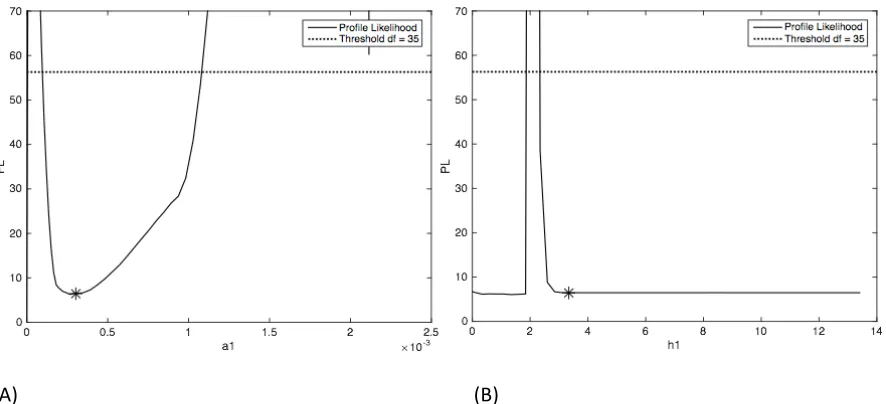
Figure 4.4: Profile Likelihood plots for assessing practical identifiability ... 45
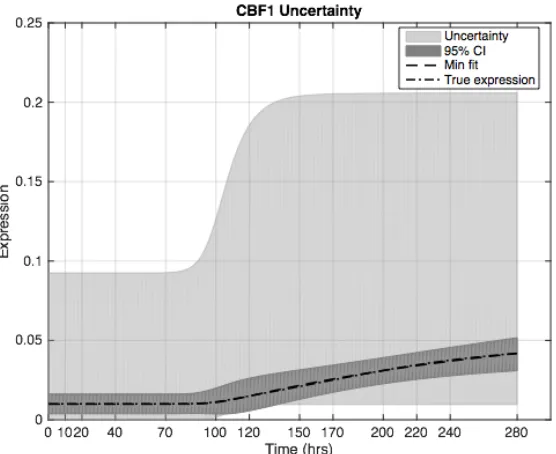
Figure 4.5: Output Uncertainty for CBF1 ... 46
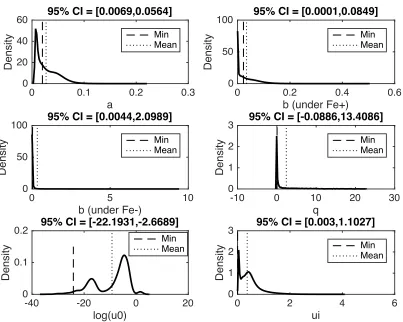
Figure 4.6: Parameter distributions for COL4 parameters: ... 50
Figure 4.7: Uncertainty for COL4 ... 52
SUPPLEMENTAL TABLES
Table S1: Comparison of frequently used UA techniques ... 122
Table S2: Comparison of commonly used SA methods ... 123
Table S3: Coefficient of Variation for the 30 parameters in the Yeast synthetic network
model in Saccharomyces cerevisiae ... 124 Table S4: Sensitivity rankings based on absolute mean elementary effects calculated by
Morris Screening and the overall rank based on the Savage scores for Yeast synthetic
network model in Saccharomyces cerevisiae ... 125 Table S5: Decay rates obtained from statistical analysis on the half-‐life experiment... 126
SUPPLEMENTAL FIGURES
Figure S1: Comparison of model Dynamics: ... 75
Figure S2: Bootstrap size selection ... 78
Figure S3: Boxplots for the 30 parameters of Yeast synthetic network model in
Saccharomyces cerevisiae ... 79
Figure S4: Uncertainty in the gene expression outputs for the 5 genes in Yeast synthetic
network model ... 83
Figure S5: Time varying sensitivity diagrams for GAL4 ... 87
Figure S6: Parameter uncertainties in the iron deficiency response model in Arabidopsis thaliana ... 88 Figure S7: Profile Likelihood plots for identifiability analysis ... 95
Figure S8: Total uncertainty in gene expressions under Fe-‐ for genes in iron deficiency
response model in Arabidopsis thaliana ... 114 Figure S9: 95% confidence intervals for gene expressions under Fe + (dark grey region) and
CHAPTER 1
Introduction
Systems Biology [1], [2] aims to understand the behavior of interacting biological system
components by gathering experimental observations and deciphering the implication of
these interactions with mathematical reasoning [3]. These observations and reasoning can
be used to understand and develop gene regulatory networks and perform metabolic
pathway analyses to explain how organisms function relative to environmental stresses or
under transgenic conditions [4]–[6].
At the genetic level, high-‐throughput experimental methods like microarray, qRT-‐PCR [7],
and RNA-‐SEQ [8], [9] are primarily employed to collect information required to develop a
gene regulatory network and to help build predictive mathematical relationships. However,
much of these experimental techniques are costly and sometimes time consuming to repeat
thus resulting in insufficient collection of data. This insufficient data poses a barrier for
researchers studying a biological system. The barrier is even greater when complex dynamic
mathematical models are used to identify and understand the interactions occurring in the
biological system.
Ordinary Differential Equations (ODEs), a type of mathematical model, are frequently
employed as dynamic models to track important inputs and their influence on transient
important to estimate to achieve the best model fit to the experimental data. The lack of
sufficient experimental data, however, leads to a condition of sparseness where the
number of model parameters exceeds the available experimental data. The parameter
values obtained under such sparse conditions can have uncertainties associated with them
that lead to inaccurate model outputs [4], [14]–[17]. If these uncertainties in parameters
are ignored, then erroneous conclusions can be inferred from the modeling results. The
need to understand and quantify the uncertainties related to mathematical models have led
to the introduction of uncertainty analysis and sensitivity analysis [18]–[20]. These
approaches have been extensively used in other areas such as risk analysis [21]–[23].
Several studies that make use of the uncertainty and sensitivity analyses in systems biology
have been reported [24]–[28]. However, these studies seem to be lacking one or more
points that might cause confusion among researchers looking to incorporate the
uncertainty and sensitivity analyses in their studies. These points include the following:
• The quantification of output uncertainty due to variability in parameters involves
two steps: parameter uncertainty quantification and forward propagation of
parameter uncertainty. Forward propagation refers to propagating the uncertainty
of the parameters by applying the uncertainty in the model equations. However, in
most of the systems biology studies, the parameter uncertainties are not calculated
differently under varied conditions, the assumed parameter uncertainties might not
be accurate thus resulting in an incorrect assessment of output uncertainties.
• A comprehensive understanding of uncertainty includes quantifying the variation in
the parameters and the outputs (i.e. uncertainty analysis/quantification),
determining the importance of parameters in relation to the change in output (i.e.
sensitivity analysis), and assessing if the parameters are identifiable (i.e.
identifiability analysis). These three analyses provide different information about
the model and the system under study. The information obtained is often
complementary and exclusive thus assisting in better understanding the model and
the system behavior under study. However, most system biology studies focus on
only one of these aspects of uncertainty that potentially results in an incomplete
understanding of the system.
• Sensitivity analysis for dynamic models include studies demonstrating the
application of variance-‐based methods [29], [30]. However, researchers have shown
that these methods are computationally intensive and infeasible for complex
systems. Screening methods, especially the method of Morris, is considered to be
an efficient alternative. However, the practical relevance of Morris Screening,
particularly to biological models, is somewhat unclear.
1.1 Objectives
• To affirm the need to conduct uncertainty analysis in the presence of data
sparseness;
• To perform the uncertainty analysis, the sensitivity analysis and the identifiability
analysis on a dynamical system model to illustrate a comprehensive understanding
of mathematical model uncertainty;
• To illustrate the use of Morris Screening in dynamic models; and
• To use the uncertainty information to develop a better understanding of iron effects
on gene expression of transcription factors in the Arabidopsis thaliana gene regulatory network
• To assess whether the parameter sensitivity analysis can be used to decrease the
sparseness in a dynamical biosystem model.
A successful completion of these analyses will provide great insight into the model behavior
and the model parameters. This information in turn will help decipher the different
biological interactions and properties of the biosystems under study.
CHAPTER 2
Background
2.1 Dynamic system models and uncertainty
There are a significant number of different models for biological systems. Broadly, these
models are classified under two types: static models and dynamic models. Although a
successful history of static models have been applied to systems biology studies [31], [32],
the use of dynamic models has a greater potential for success. The reason for greater
success is that a dynamic model can provide the systems behavior over time, unlike static
models that provide information only at a particular state [33]. However, dynamic models
are limited and typically include a large number of parameters that need to be estimated
with sparse data. Under such sparse conditions, large uncertainties are associated with the
parameters as well as the predictions. These uncertainties could lead to incorrect inferences
about the system. To ensure better understanding of the system, the uncertainties in the
dynamic models must be understood properly. However, studies that perform a
comprehensive assessment of uncertainties in dynamic models are lacking.
2.2 Uncertainty Analysis
Parameter uncertainty analysis is important in the evaluation of any mathematical model
and critically important when these models are evaluated with sparse data sets. Uncertainty
Analysis (UA) consists of two components: parameter uncertainty quantification and
the uncertainty would be to initially assess the parameter uncertainty and then propagate
this uncertainty into the model to determine the model output uncertainty. Yet, most of the
uncertainty analysis performed in systems biology have only utilized forward propagation
based on some form of random sampling [35]. The parameter uncertainty in these studies is
either based on a priori knowledge or is assumed. Such assessments are not accurate
because system behavior and properties vary for different conditions. Several uncertainty
analysis methods frequently used in systems biology studies as well as in other disciplines
are listed in Table 2.1. The table summarizes the type of results obtained, the advantages
and the limitations of the uncertainty analysis methods. In the sections following, these
methods are discussed based on the findings from previous studies. Moreover, the
advantages and applicability of one of the uncertainty analysis methods, Bootstrap method
used in this study, is highlighted.
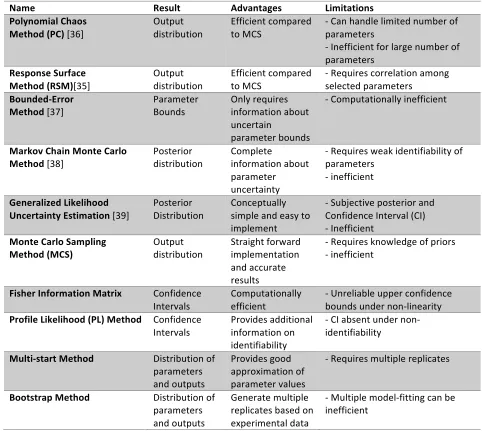
Table 2.1: Comparison of frequently used UA techniques
Name Result Advantages Limitations
Polynomial Chaos
Method (PC) [36] Output distribution Efficient compared to MCS -‐ Can handle limited number of parameters -‐ Inefficient for large number of parameters
Response Surface
Method (RSM)[35] Output distribution Efficient compared to MCS -‐ Requires correlation among selected parameters
Bounded-‐Error
Method [37] Parameter Bounds Only requires information about uncertain
parameter bounds
-‐ Computationally inefficient
Markov Chain Monte Carlo
Method [38] Posterior distribution Complete information about parameter uncertainty
-‐ Requires weak identifiability of parameters
-‐ inefficient
Generalized Likelihood
Uncertainty Estimation [39] Posterior Distribution Conceptually simple and easy to implement
-‐ Subjective posterior and Confidence Interval (CI) -‐ Inefficient
Monte Carlo Sampling
Method (MCS) Output distribution Straight forward implementation and accurate results
-‐ Requires knowledge of priors -‐ inefficient
Fisher Information Matrix Confidence Intervals
Computationally efficient
-‐ Unreliable upper confidence bounds under non-‐linearity
Profile Likelihood (PL) Method Confidence Intervals
Provides additional information on identifiability
-‐ CI absent under non-‐ identifiability
Multi-‐start Method Distribution of parameters and outputs
Provides good approximation of parameter values
-‐ Requires multiple replicates
Bootstrap Method Distribution of parameters and outputs
Generate multiple replicates based on experimental data
-‐ Multiple model-‐fitting can be inefficient
2.2.1 Non-‐sampling based Uncertainty Analysis
Polynomial Chaos (PC) method and Response Surface Method (RSM) [35] are two popular
non-‐sampling based methods. PC, based on the concept of Wiener [36], uses spectral
on applying and improving PC methods. These studies show that PC methods are efficient
compared to Monte-‐Carlo methods, yet in the presence of large number of uncertainties,
these methods become inefficient. Debusschere et al. [47] discuss the numerical challenges
of applying PC methods and showed that high PC orders are necessary to avoid negative
values for strictly non-‐negative parameters and truncation errors at a high computational
cost. RSM [35] develops a response surface using few selected parameters to describe the
relationship between parameters and output. RSMs work better when they are highly
correlated but may not be able to detect important interactions when two parameters are
super-‐additive [48]. Both, PC and RSM are forward propagating uncertainty methods.
Another, non-‐sampling based method is the bound-‐error method [37]. Bound-‐error method
is based on interval analysis method which does not require probability distribution
information of the uncertain parameters. It rather assumes the bounds of the parameters
to quantify uncertainty. Several works have used this method and improvements have been
put forward [49]–[51]. Yet, the interval analysis based bound-‐error methods can still be
computationally infeasible for complex biological models.
2.2.2 Sampling based Uncertainty Analysis
Sampling based uncertainty analysis falls under two approaches: Bayesian approach and
Frequentist approach. Bayesian methods are probabilistic approaches based on Bayesian
theory while the frequentist approaches are based on multiple model fitting [52]. Most
(GLUE) of Beven and Binley [39] and Markov Chain Monte Carlo (MCMC) [38] methods.
GLUE and MCMC result in posterior probability that expresses the parameter uncertainty.
However, the posteriors obtained from GLUE are subjective and may not be the true
parameter uncertainty distribution. Like all Bayesian methods, both GLUE and MCMC
require the knowledge of priors [53]. Vrugt et al. [54] compared GLUE, and two MCMC
based approaches, Differential Evolution Adaptive Metropolis (DREAM) [55] and Delayed
Rejection Adaptive Metropolis (DRAM) [56].They observed that GLUE resulted in similar
estimates of parameter and prediction uncertainty as MCMC approaches. However, MCMC
based DREAM was much more efficient with regards to number of model evaluations
compared to GLUE and DRAM. According to Iorgulescu et al. [57], GLUE may require billions
of model evaluations to generate only a few good solutions. Though, DREAM MCMC is
efficient in comparison with GLUE and DRAM, it can still be inefficient and infeasible for
complex and stiff biosystem models [58]. Yang et al. [48] pointed out the difficulty in
constructing a likelihood function and the difficulty in assessing multi-‐modal distributions
with MCMC approaches.
Methods that fall under the frequentist approach are the Fisher Information Matrix (FIM),
the Profile Likelihood method (PL), the Multi-‐start method and the Bootstrapping method.
These methods result in the confidence intervals (CIs)of the parameters. Multi-‐start and the
bootstrap result in parameter distributions [59]–[62]. Studies have shown that the
parameters are identifiable [63], [64]. However, Joshi et al. [62] showed that FIM has two
important shortcomings: 1) it only provides lower bounds when the model is non-‐linear and
2) the confidence interval is symmetric which may not be true in all models. In addition, FIM
is a parameter dependent measure and requires prior knowledge about the parameters
[65].
The PL approach provides information about identifiability of parameters [16], [66], [67].
Multi-‐start methods can be used along with bootstrap methods to quantify parameter
uncertainties. Bootstrap methods are able to generate multiple samples based on few
experimental observations. These samples are used to quantify the parameter uncertainties
and subsequently the output uncertainties thus making them suitable for systems biology
studies. However, multiple model fitting of bootstrap samples can be computationally
expensive for complex models.
2.3 Sensitivity Analysis
Sensitivity Analysis (SA) quantifies the impact of parameter uncertainties on the model
outputs [68]. The objective of SA is to identify critical inputs (parameters and initial
conditions) of a model. Some of the important and frequently used SA methods are
compared in Table 2.3. The review below reports the findings of several SA studies to
establish the appropriate SA method for dynamic systems.
Table 2.3: Comparison of commonly used SA methods
Name Type Advantages Disadvantages
1. Local SA Local -‐ Easy
implementation -‐ Low computational cost
-‐ Non-‐generalizable results
-‐ Unable to detect parameter interactions 2. Global SA
i. Partial Rank Correlation Coefficient (PRCC) [69]
ii. Standardized Rank Regression
Coefficient (SRCC) [69]
Correlation/Regression based
-‐ Computationally efficient
-‐ Unreliable under non-‐monotonicity
iii. Sobol Method [70]
iv. Extended Fourier Amplitude
Sensitivity Test [71]
Variance-‐based -‐ Model independent -‐ Quantitative
measure of sensitivity
-‐ Computationally inefficient
v. Morris Screening
[72] Screening method -‐ Computationally efficient -‐ Comparable to Sobol indices
-‐ Provides qualitative information only
2.3.1 Local Sensitivity Analysis
Research on local SA studied the effect of small perturbations of parameters, typically about
the mean value, on the model output. The application of local SA to signal transduction and
metabolic pathway models ([73], [74]) report the ease in implementation and the low
computational time required. However, these methods are not able to study the effects of
simultaneous parameter variations and the sensitivity is investigated in the immediate
parameter interactions and are not generalizable over the entire parameter range. A list of
commonly used local SA methods can be found in Hamby [75].
2.3.2 Global Sensitivity Analysis
In contrast to local SA, global SA considers the entire parameter range, facilitates the
simultaneous variation of parameter values, and allows the exploration of parameter
interaction effects [68]. There are three Global SA methods: correlation and regression
methods, variance-‐based methods, and screening methods. Partial Rank Correlation
Coefficient (PRCC) [69], and Standardized Rank Regression Coefficient (SRRC) are the most
efficient and reliable correlation and regression methods [76]. However, these methods are
suitable for monotonic parameter-‐output relationships and are not reliable under non-‐
monotonicity that are usually present in biological models. Variance-‐based methods provide
information about the fractional variance from individual parameters and groups of
parameters on the output variance. They do not depend on assumptions of linearity or
monotonicity which make them suitable for biological models [30]. The method of Sobol
[70] and the extended Fourier amplitude sensitivity test (eFAST) [71] are the two most
commonly used variance-‐based methods. The method of Sobol is relatively easy to
implement compared to other variance-‐based methods [77] and its modified form is
efficient as the eFAST method [78]. Zheng & Rundell [29] compared sensitivity results of the
local SA, the PRCC, the method of Sobol and the eFAST methods on a model of the ErK-‐
and the global SA. Interestingly, the PRCC produced consistent results as the Sobol and the
eFAST method suggesting the robustness of the sensitivity pattern of the model to the
monotonic assumption. A comparison of the PRCC and the eFAST method on different
deterministic and stochastic biological models by Marino et al. [30] revealed differences
between the PRCC and eFAST results under non-‐linear and non-‐monotonic relationships. As
PRCC performs well under monotonicity, its results could be misleading under non-‐
monotonic and non-‐linear conditions. Overall, the variance-‐based methods can be
impractical to use if the model is very complex and the number of parameters is large [79].
An efficient alternative to the variance-‐based methods are the screening methods. The
trade-‐off, however, is that screening methods only provide qualitative information i.e. the
rank of the parameters in terms of sensitivity. Morris Screening [72] is a commonly used
screening technique since it considers the entire parameter space and allows variation of
multiple parameters unlike other screening methods like iterated fractional factorial design
(IFFD) [80], sequential bifurcation (SB) [81] and Cotter’s design [82]. An extension to the
Morris method was suggested by Campolongo et al. [83] to improve the sensitivity
assessment during non-‐monotonicity. The study found that the new absolute mean of
elementary effects was comparable to the total sensitivity index from the method of Sobol.
Sensitivity analyses for dynamic models should be able to consider the entire dynamics.
Focusing at a particular time point of interest, as done in most of the existing studies in the
period. Some studies have been performed recently to include the dynamical aspect in SA
of the system [29], [30], [74], [84] using the PRCC, the Sobol and the eFAST methods. But,
similar studies using Morris screening is lacking and is part of the current study.
2.4 Preliminary Iron deficiency response model in Arabidopsis thaliana
A successful study that can explain the various interactions occurring under dynamic iron
conditions could be very important since iron is required for the growth and development
of plants. Such studies could help develop ways to sustain the plant’s growth and
productivity under iron deficient conditions. This kind of study has been of great interest in
biology and more so in the light of systems understanding through gene regulatory network
construction and gene interactions modeling. Several efforts have been performed to study
iron responses in Arabidopsis thaliana [85]–[92]. These responses are known to be
transcriptionally induced by transcription factors. A set of transcription factors (TFs) COL4,
ETF9, ASIL2, and MYB55 that influence known iron response TFs were identified by
Koryachko et al. [85]. Additional TFs bHLH34, bHLH104, and bHLH05 (ILR3) were identified
by Li et al. [90] and Zhang et al. [91]. Koryachko et al. (yet to be published) incorporated
these TFs and their targets to formulate a gene regulatory network (Figure 1) and an
Ordinary Differential Equation (ODE) based iron deficiency response model to describe the
interactions occurring under iron deficient conditions in Arabidopsis thaliana. The iron deficiency model is still under study and the ODE-‐based equations used in this study is a
parameters and the outputs could help in better understanding the iron responses in
Arabidopsis thaliana.
Figure 1: Iron Deficiency Gene Regulatory Network in Arabidopsis thaliana: Top row shows the TFs and the bottom row shows their targets. Lines with arrow-‐head indicate activation while lines with flat end indicate inhibition. Bold lines indicate direct binding between regulator's protein and target's promoter.
CHAPTER 3
Methods
In the previous chapter, various uncertainty and sensitivity analysis techniques were
discussed. This chapter describes, in detail, the methods used for this study that are
applicable for a comprehensive understanding of uncertainty for any complex dynamic
biological model under sparse data conditions.
3.1 Model
The methods described in this section were first applied to a Yeast synthetic network model
in Saccharomyces cerevisiae and then to the preliminary iron deficiency model in Arabidopsis thaliana.
3.1.1 Yeast Synthetic Network Model in Saccharomyces cerevisiae
The synthetic network model used in this study is based on the synthetic network in the
yeast Saccharomyces cerevisiae built by Cantone et al. [93]. Figure 2 displays the synthetic network. The network consists of five interacting genes. Cantone et al. described the
network using a system of nonlinear Delay Differential Equations (DDEs) and hill kinetics
[93]. The variables in the model represent the mRNA abundances of each gene and the
degradation kinetics were assumed first-‐order. In our study, we made two changes to the
original DDE based model. The delay function and the rectangular window function were
simplify the model and to modify the model into an Ordinary Differential Equation (ODE)
based system of equations like the preliminary iron deficiency model in Arabidopsis thaliana. The modification to the yeast Saccharomyces cerevisiae model, however, did not change the dynamics of the system and the output expression as shown in supplemental
section (Figure S1).
Following these changes, the rate of change of the mRNA concentrations of the genes in the
yeast synthetic network (Figure 2) were expressed as:
𝑑𝑥𝑑𝑡# = 𝛼# + 𝑣# 𝑥)
*+
𝑘#*++ 𝑥)*+ 1 +𝑥.*/ 𝑘0*/
− 𝑑#𝑥# ∙ 𝑡3+
𝑐03++ 𝑡3+ (1)
𝑑𝑥𝑑𝑡0 = 𝛼0+ 𝑣0 𝑥# *6
𝑘)*6+ 𝑥#*6 − 𝑑0− 𝛽#∙
𝑐)3+
𝑐)3++ 𝑡3+ ∙ 𝑥0 (2)
𝑑𝑥)
𝑑𝑡 = 𝛼)+ 𝑣)
𝑥0*8
𝑘9*8 + 𝑥
0*8 1 + 𝑥99 𝛾99
− 𝑑)𝑥) (3)
𝑑𝑥𝑑𝑡9 = 𝛼9+ 𝑣9 𝑥) *;
𝑘.*; + 𝑥 )*;
− 𝑑9− 𝛽0∙ 𝑐) 3+
𝑐)3+ + 𝑡3+ ∙ 𝑥9 (4)
𝑑𝑥.
𝑑𝑡 = 𝛼.+ 𝑣.
𝑥)*< 𝑘=*<+ 𝑥
)*<
− 𝑑.𝑥. (5)
where [CBF1] = 𝑥#; [GAL4] = 𝑥0; [SWI5] = 𝑥); [GAL80] = 𝑥9; [ASH1] = 𝑥., 𝑑>, 𝑖 = 1, . . . , 5 are
basal activities, 𝑣> represent the maximal transcription rates, 𝛾 is an affinity constant.
𝑐#, 𝑐0, 𝑐) are constants with value 16, 100 and 10 respectively, which are used to model the
100 minutes delay in the HO promoter binding for CBF1 gene and the 10 minutes transient
increase in mRNA stability due to experimental washing steps. The concentrations are
reported in arbitrary units [a.u.], the degradation rates 𝑑#, 𝑑0, 𝑑), 𝑑9, 𝑑. in 𝑚𝑖𝑛E# , the
Michaelis-‐Menten constants 𝑘#, 𝑘0, 𝑘), 𝑘9, 𝑘., 𝑘= in [𝑎. 𝑢.], the affinity constant in [𝑎. 𝑢.],
the basal activities 𝛼#, 𝛼0, 𝛼), 𝛼9, 𝛼. in [𝑎. 𝑢. 𝑚𝑖𝑛E#], the maximal transcription rates in
[𝑎. 𝑢. 𝑚𝑖𝑛E#], and the coefficients 𝛽
# and 𝛽0 for the magnitude of the increase in mRNA stability in [𝑚𝑖𝑛E#].
Figure 2: Yeast Synthetic Network in Saccharomyces cerevisiae [93]: Lines with arrow heads indicate activation while lines with flat ends indicate inhibition
3.1.2 Preliminary Iron Deficiency Response Model in Arabidopsis thaliana
The preliminary iron deficiency response model is based on the gene regulatory network in
Arabidopsis thaliana that consists of 6 regulator genes and their 7 target genes (Figure 1). For our study, we focused on the 6 regulators in the GRN. Two set of ODEs were used to
describe the gene expression under iron sufficient (Fe+) and iron depleted (Fe-‐) conditions
which are:
𝑑𝑥
𝑑𝑡 = 𝑎 − 𝑏𝑥 (6)
𝑑𝑥𝑑𝑡 = 𝑎 + 𝑞 𝑢> 1 1 + 𝑢>
𝑢K− 1 𝑒
EMNO − 𝑏𝑥 (7)
where 𝑥 represents the gene expression, 𝑎 the constitutive transcription rate, 𝑏 the
degradation rate, 𝑞 represents the sensitivity of iron on gene expression, 𝑢> represents the
rate of rise in iron signal and 𝑢K represents the delay in the iron effect. Equation 6 describes
the Fe+ condition and (7) describes the Fe-‐ condition.
3.2 Bootstrap Sampling for Data Generation
Due to the high expense and the long duration associated with high-‐throughput methods
like microarrays, qRT-‐PCR, and RNA-‐Seq used to measure gene expressions, the number of
data replicates are limited. Lack of multiple data replicates make it challenging to quantify
the parameter distribution. However, a bootstrap method is able to generate such
replicates required to characterize the parameter distribution [62]. We used the parametric
mean expression to create data samples. The mean expression and the sample were
created as follows:
(i) Mean expression: The parameter values provided in [93] were used as the true values for the yeast synthetic model parameters in this study. These values were
substituted in the model equations (1) – (5) to obtain the mean expression. To
create data sparseness, expression values at 13 time-‐points: 0, 10, 20, 40, 70,
100, 120, 150, 170, 200, 220, 240 and 280 hours were taken. For the iron
deficiency response model, however, the 3 qRT-‐PCR measurements at 4
different time-‐points each for Fe+ and Fe-‐ conditions were available. The mean
expression was the average of these measurements.
(ii) Parametric Bootstrap sample generation: Once the mean expressions were calculated, the next step involved generation of noise samples. Parametric
bootstrap creates random noise based on the information on the distribution of
the data. As a general practice, we considered a Gaussian distribution. A set of
random Gaussian noise were created. These noises were added to the mean
expression to get the bootstrap data samples. In case of the yeast synthetic
model, the variance of the Gaussian noise was selected such that the noise to
signal ratio was 10%. For the iron deficiency model, the variances of the
measurements were used to generate the Gaussian noise. Statistically, an
estimate of variance based on 3 samples may not be truly reflective of the
considered those variances as approximate estimations. More than 20,000
bootstrap samples were generated using this approach. The resulting samples
were freed of any outliers using the Tukey’s outlier analysis [94]. Out of the
20,000 samples, 2,500 random bootstrap samples were chosen for further
analysis. The appropriate size of the bootstrap i.e. 2,500 was selected
considering that the mean values for the parameters did not change for any
greater bootstrap size (Figure S2 in Supplemental section).
3.3 Parameter Estimation
Parameter values were estimated by fitting the model to the bootstrap generated sample
data. For model fitting, a local optimization routine “fmincon” was employed in MATLAB. A
cost function based on weighted sum of squares were defined for both models. The
weighted sum of squares cost function was defined as:
𝐽 = (𝑦3ST3
> 𝑗 − 𝑦
UVW> (𝑗))0 𝜎>@0
Z
@[# \
>[#
(8)
where 𝑗 denotes the time point and 𝑖 represents the gene.
3.4 Uncertainty Analysis
Uncertainty analysis involves quantifying the parameter uncertainty and the uncertainty in
the model outputs resulting from these parameter uncertainties. To quantify the parameter
uncertainty, model-‐fitting was performed on each of the 2,500 bootstrap sample data that
respective distributions were constructed for each parameter. Next, the output
uncertainties were characterized using forward propagation of the parameter uncertainty.
In forward propagation, the 2,500 parameter combinations were applied to the model to
generate the output distribution. Finally, confidence intervals were calculated for the
parameters as well as the outputs.
3.5 Identifiability Analysis
Identifiability denotes the ambiguity in the estimation of parameters [95]. A parameter is
non-‐identifiable if it cannot be estimated with a finite confidence interval. Two types of
non-‐identifiability occur in a model, namely structural non-‐identifiability and practical non-‐
identifiability [16]. Structural non-‐identifiability, as the name suggests, is related to the
structure of the model while practical non-‐identifiability is related to the quality and
amount of data. Since, the focus of our study was on sparseness of data, we limited our
identifiability tests to the practical non-‐identifiablity of the parameters. We used the Profile
Likelihood approach [16] to assess the practical non-‐identifiability of the parameters in the
two models.
Profile Likelihood approach detects identifiability by assessing the flatness of likelihood. If
the cost function is defined as 𝜒0, then the profile likelihood for a particular parameter 𝜃 > is:
𝜒_`0 𝜃> = mind
efN 𝜒 0(𝜃)
(9)
As shown in Equation 9, the cost function is re-‐optimized with respect to all other
identifiability of the parameters, a threshold is defined. The threshold corresponds to the
inverse chi-‐squared value for a confidence 𝛼 and degree of freedom equal to the total
number of parameters. The profile likelihood for the parameters are plotted and checked if
they intersect the threshold at either side of the minimum cost function. A parameter is
considered practically non-‐identifiable if its profile likelihood flattens out at either end (i.e.
the profile likelihood does not intersect the threshold at either end).
3.6 Sensitivity Analysis
Sensitivity analysis quantifies the change in output due to the change in the parameters. For
our study, we chose the Morris Screening method, one of the screening methods described
earlier for sensitivity analysis. Morris Screening method [72], like any other screening
method, is based on One-‐at-‐a-‐time (OAT) designs. These designs are local methods as they
evaluate the effects of varying only a single parameter around its nominal values at a time
on the outputs. However, unlike most OAT designs, Morris Screening calculates sensitivities
at multiple points in the parameter space thus making it a global method [96].
Morris defines the elementary effect for determining the sensitivity of the parameters. For
a model with 𝑘 parameters, the region of experimentation i.e. the entire parameter space is
denoted by 𝜔, which is a regular 𝑘 -‐dimensional 𝑝 -‐ level grid. In this grid, each parameter
𝑥> may take values from {0, 1/(p-‐1), 2/(p-‐1), . . . ,1}. For a vector of values 𝑥, the elementary
𝐸𝐸> 𝑥 = 𝑦 𝑥#, … , 𝑥>E#, 𝑥> + ∆, 𝑥>l#, … , 𝑥m − 𝑦(𝑋)
∆ (10)
where ∆ is a multiple of 1/(𝑝 − 1) and 𝑋 is such that 𝑋 + ∆ is still in the region of
experimentation. A total of 𝑟 elementary effects were calculated. Morris calculates the
mean, 𝜇, and standard deviation, 𝜎, of the 𝑟 EEs to indicate the sensitivity. However, under
non-‐monotonicity, it was found that the mean underestimated the sensitivities [83]. In our
study, we calculated the absolute mean values as suggested in [83] to represent the
sensitivities. The absolute mean indicates the overall effect of a parameter on the output
while the standard deviation indicates the effect due to interactions with other parameters.
The vector of parameter values used to calculate the EEs were sampled based on radial
points described by Campolongo et al. [97].
3.6.1 Extension to Morris Method
Studies that have applied Morris Screening method on dynamic models are rare. In the few
studies where the method has been applied to a dynamic model, sensitivity is defined at a
specific time point of interest. This approach cannot provide the time-‐varying nature of
parameter sensitivities. In our study, we calculated the sensitivities at all time-‐points to
examine the time-‐varying sensitivities. Moreover, for multiple outputs, Morris Screening
provides multiple rankings and is difficult to understand how a parameter ranks overall. So,
to get an overall rank, we used the Savage Scores [98]. Savage Scores were calculated as
𝑆> = 1 𝑗 \
@[#
(11)
where 𝑖 is the rank assigned to the 𝑖th order statistic in a sample of size 𝑛.
CHAPTER 4
Journal Article (IEEE Transactions on Systems, Man, and Cybernetics: Systems)
Uncertainty and Sensitivity Analysis in Dynamic Biological Systems under Data Sparseness
Basnet, L., Koryachko, A., Matthiadis, A., Muhammad, D., Tuck, J., Williams, C.M., Long, T., Ducoste, J.J.
Abstract
A comprehensive understanding of the uncertainties due to data sparseness in dynamic
models used in systems biology studies can prevent misleading deductions and provide
valuable biological information about their systems. Given the scope and flexibility of
Ordinary Differential Equation (ODE) based dynamic models in biological studies,
comprehensive uncertainty studies for these models are lacking. In this research, two ODE-‐
based dynamical models: the yeast synthetic network model in Saccharomyces cerevisiae and the preliminary iron deficiency response model in Arabidopsis thaliana were studied under data sparseness. The goal was to quantify the variability in the model parameters and
outputs, assess the identifiability of parameters and identify the importance of parameters
to deduce important biological information. Bootstrap results showed high uncertainties in
parameters and gene expressions in the yeast synthetic network model. Practical
identifiability assessment using profile likelihood suggested high data sparseness for both
models. Morris screening sensitivity results explained the regulatory effects of SWI5 and
ASH1 genes on CBF1 in the yeast synthetic network model. Clear influence of iron effect in
COL4 gene expression, and of constitutive transcription and degradation in bHLH104 gene
expression were identified in the Arabidopsis thaliana gene regulatory network. Keywords
Systems biology; Dynamic model; Data Sparseness; Uncertainty; Identifiability; Sensitivity;
4.1 Introduction
Systems Biology [1], [2] aims to understand the behavior of interacting biological system
components by gathering experimental observations and deciphering the implication of
these interactions with mathematical reasoning [3]. These observations and reasoning can
be used to understand and develop gene regulatory networks and perform metabolic
pathway analyses to explain how organisms function relative to environmental stresses or
under transgenic conditions [4]–[6].
At the genetic level, high-‐throughput experimental methods like microarray, qRT-‐PCR [7],
and RNA-‐SEQ [8], [9] are primarily employed to collect information required to develop a
gene regulatory network and to help build predictive mathematical relationships. However,


![Figure
2:
Yeast
Synthetic
Network
in
Saccharomyces
cerevisiae
[93]:
Lines
with
arrow
heads
indicate
activation
while
lines
with
flat
ends
indicate
inhibition](https://thumb-us.123doks.com/thumbv2/123dok_us/1769155.1227674/31.612.217.415.335.544/synthetic-network-saccharomyces-cerevisiae-indicate-activation-indicate-inhibition.webp)

![Figure
4.2:
Yeast
Synthetic
Network
in
Saccharomyces
cerevisiae
[93]:
Lines
with
arrow
heads
indicate
activation
while
lines
with
flat
ends
indicate
inhibition](https://thumb-us.123doks.com/thumbv2/123dok_us/1769155.1227674/49.612.179.530.70.246/synthetic-network-saccharomyces-cerevisiae-indicate-activation-indicate-inhibition.webp)