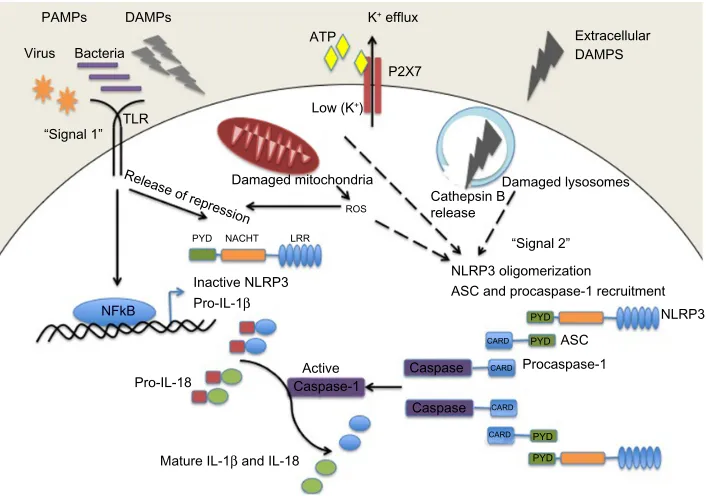
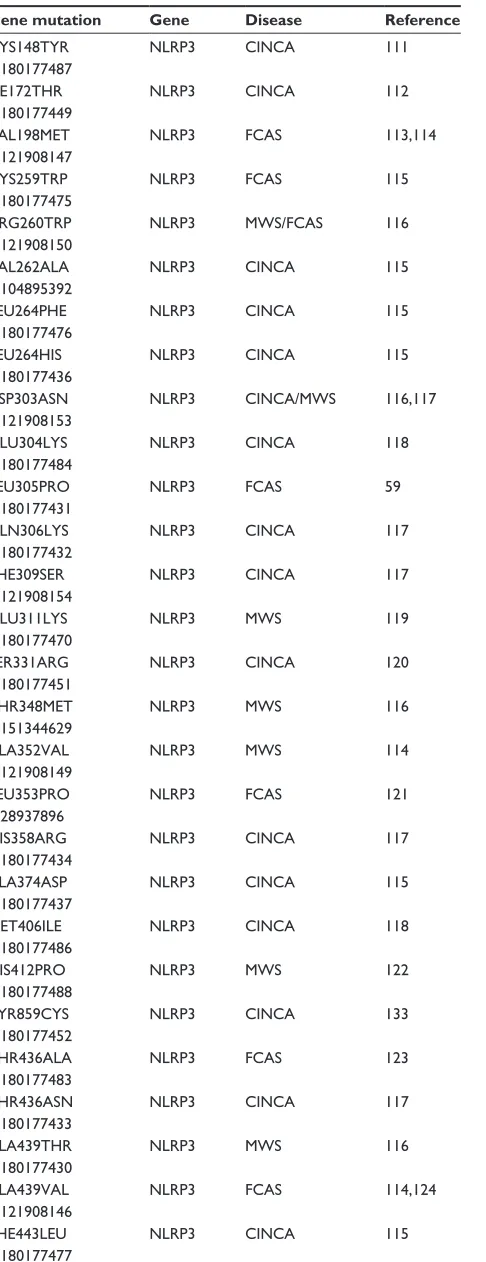
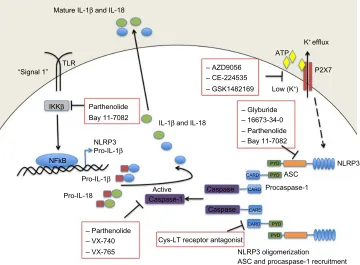
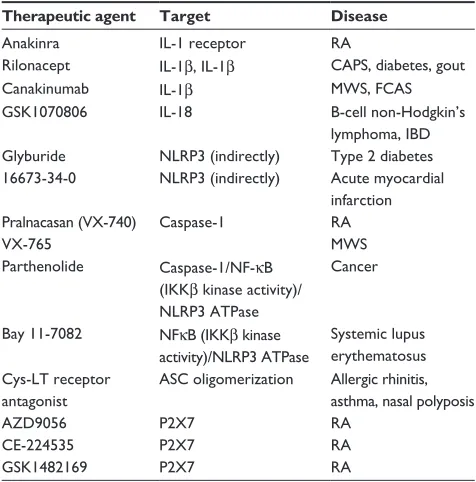
Targeting the NLRP3 inflammasome in chronic inflammatory diseases: current perspectives
Full text
Figure

Related documents
Designing of low energy manufacturing process is key of sustainable development in a high energy intensive plant like steel rolling process The hot charging
Severity: The behavior in question must occur more frequently in the child than in other children at the same developmental stage. Early onset: At least some of
This specific problem seeks to minimize total cost by simultaneously locating the distribution centers and designing the vehicle routes that satisfy pickup and
In the present study, we present an innovative SPECT diagnostic tool for abdominal aortic aneurysm (AAA) produced from injectable polysaccharide microparticles radiolabeled
the relevance of antitrust law to the noneconomic concerns surrounding media concentration: "[S]uppose that a single wealthy family were to acquire the leading
Three dissenting opinions were voiced in Bodzin which implied that home office deductions should not be allowed the liberal treat- ment given by the majority. Judge
Aquinas says that all those things that practical reason naturally understands to be good, that is to say, all those things to which we have a
11: Apoptosis analysis by FCM using AnnexinV-FITC/PI staining on the HT-29 cells treated by synthesized compounds; Group-II: Tumour control without compound and
