iMAGA: INTRON MULTIPLE
ALIGNMENT USING GENETIC
ALGORITHM
V. AMOUDACentre for Bioinformatics
Pondicherry University, Puducherry – 605014, India V. SELVARAJ
Centre for Bioinformatics
Pondicherry University, Puducherry – 605014, India S. KUPPUSWAMI
Department of Computer Science
Pondicherry University, Puducherry, Puducherry – 605014, India S.BUVANESWARI
Centre for Bioinformatics,
Pondicherry University, Puducherry – 605014, India Abstract :
Multiple sequence alignment (MSA) is one of the multi-dimensional problems in biology. This paper describes a new approach to solve MSA, a NP-hard problem using modified Genetic Algorithm with new mutation operator. A web based tool iMAGA (Intron Multiple Alignment using Genetic Algorithm) is developed for aligning the intron sequences in order to find the pattern. It has two modules (i) iExtractor/ iClassifier which extracts and classifies introns (ii) iAligner/ Pattern Finder which aligns the intron sequences and finds the patterns.
iAligner, the core module of the tool aligns any type of sequences (DNA, RNA, Protein & Intron). In this module GA is applied in which the chromosome consists of gap positions. On applying conventional mutation operator leads to problems like repetition and increase in number of gap positions. To overcome these problems, a newly designed mutation operator X-Shuffler is proposed.
To validate the alignment, the sum-of-pairs score is used to compare iMAGA with widely used tools. The data sets chosen from the standard BaliBASE, SMART and OXBENCH benchmark alignment suite.
To validate the pattern obtained using iMAGA tool, the similarity percentage of the pattern is compared with MEME, a widely used motif finder. Dataset of Saccharomyces Cerviceae with 254 intron sequences are used to prove this work. The tool is available at www.imaga.bicpu.edu.in.
Keywords: Genetic algorithm; mutation operator; multiple sequence alignment. 1. Introduction
Multiple sequence alignment is an essential pre-requisite in molecular sequences analysis. This has lead to the development of different software tools. MSA can be used in Phylogenetic analysis to trace the path of evolution. The most general purpose of multiple sequence alignment is to find highly conserved region or embedded patterns. Patterns / Motifs are well conserved regions of sequence generally organized around one or two very highly conserved residues.
Genetic algorithm (GA) is a search technique used to optimization and search problems, imitating the patterns of genetic reproduction in living organism. Its flexibility in algorithm, in assigning the fitness function and its efficiency in solving NP-hard problems made us to choose GA to solve MSA.
2. Multiple Sequence Alignment (MSA)
Multiple Sequence Alignment (MSA) is identified as one of the challenging tasks in bioinformatics belongs to a class of hard optimization problems called combinatorial problems [8]. Multiple sequence alignment allows comparison of sequences by simultaneously aligning set of sequences.
The main problem in MSA is its exponential complexity with the considered input data set. These alignments may be used to identify profiles or hidden models that may be used to acquire knowledge for distantly related members of the family sequences, newly discovered sequences, and existing sequence databases.
To align multiple sequences with reasonable computer resources, a number of methods have been developed. Many of the methods are heuristics ones which attempt to find good alignment that are not necessarily optimal. The heuristic approaches for multiple sequence alignment include the regional approaches, the tree-based approaches, the consensus approaches, the random graph based approaches.
There are many methods proposed to predict patterns which includes (MEME) Multiple Em for Motif Elicitation), Gibbs sampler. The later has the advantage of spending lower computation time where as former one has the advantage of prediction accuracy with the disadvantage of more computation time. In this tool a new approach, based on genetic algorithm is used to align the intron sequences to find the pattern. The predicted results obtained by the tool are more accurate and require less computation time than the MEME tool.
2.1. Genetic Algorithm
Genetic Algorithms[19] are search techniques used to optimize and search problems, imitating the patterns of genetic reproduction in living organism. GAs is implementing by computer simulation in which successive modifications of a collection referred as population of parameter combinations in the search space. Population
of abstract representations (called chromosomes) of candidate solutions (called individuals) to an optimization
problem evolves toward better solutions.
They are designed to satisfy the four basic evolutionary conditions. • The ability for an individual to reproduce itself.
• The existence of a self – reproducing population. • The existence of variety among the individuals and • The survival ability associated with the variety [12].
The developed tool iMAGA uses GA for the alignment of intron sequences. The patterns are identified from the alignment.
2.2.iMAGA (intron Multiple Alignment using Genetic Algorithm)
Various MSA tools available to align protein and DNA sequences can also be used for intron sequences. The proposed tool is developed specially for aligning the intron sequences where protein/ DNA sequences can also be used.
The intron sequences required for the alignment is extracted and classified using a separate module developed in the tool. The core module of tool is to align the sequences using genetic algorithm and to identify the pattern.
2.2.1. Why intron
Recent research shows that introns are "a complex mix of different DNA, much of which are vital to the life of the cell." As their functions are being determined, the relationship of introns to cancer and their use as tumor markers is also being explored. Several functions for introns have already been identified, and evidence for a role for them is indicated by the finding that some intron alterations are directly related to the development of cancer [25].
3. Method
The developed tool iMAGA consists of two modules.
3.1. Module I - iExtractor / iClassifier
3.1.1. iExtractor
The input for the extraction of introns can be given by any of the following:
1. The sequences can be imported from the Genbank by giving the appropriate accession number. 2. Paste the appropriate sequence in Genbank format in the text box provided.
3. Browse the text file containing the sequences
The given input sequences will be uploaded directly into the module 1 to extract the introns. A special sub-program in Biojava (Feature extraction) is used for the extraction process. The related information of the introns are displayed after the extraction.
3.1.2. iClassifier
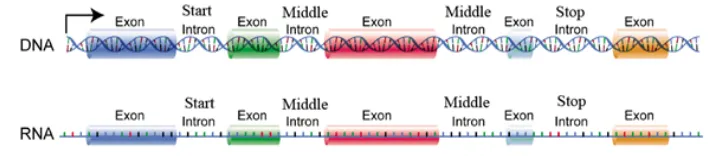
This module classifies the introns based on the nucleotide positions as start intron, middle intron and stop intron. It is evident that intial intron hold the regulatory signals such as regulators, enhancers and promoters which are used as tumor markers and also used for cancer studies[25].
Fig. 1: Classification of Introns
The classified intron sequences are displayed in FASTA format with accession number, organism, definition, location, length and it is used as input for the alignment. The hyperlink is also provided to download the extracted intron sequences.
3.2. Module II: iAligner/ PatternFinder
This module is used for (i) Multiple Sequence Alignment (MSA) (ii) to find pattern from the alignment.
3. 2. 1. iAligner
This module aligns the given input sequences using proposed genetic algorithm with a newly designed operator called X-Shuffler mutation operator. The values for parameters like Population size (50- 1000), no. of generations (1-5) and crossover type (1, 2) are assigned by the choice of the user.
3. 2. 1. 1. Proposed GA with new operator to MSA
Genetic algorithms [5], [20] are stochastic approaches based on the concept of biological evolution and biological genetics. The methods available in applying Genetic Algorithm (GA) to Multiple Sequence Alignment [13] are without gaps (local) and with gaps (global). In this work, MSA with gap was used for getting the global alignment.
iMAGA with the proposed algorithm operates on chromosome that encodes possible solutions of the problem. The crossover and mutation operators are applied to generate new chromo-somes in the search space.
3. 2. 1. 2. Selection
Selection operator determines which chromosome survives in each generation. In this process a combination of Roulette wheel selection and Elitism is implemented.
Initial population is generated with number of chromosomes specified by the user. The required numbers of chromosomes are selected randomly using the Roulette Wheel selection operator to perform crossover and mutation operation.
3. 2. 1. 3. Fitness
The sum-of-pairs function [18] is used to evaluate the fitness of chromosomes. The corresponding alignment of each chromosome is generated to calculate the fitness value. The sum-of-pairs score is defined as the sum of pairwise scores of all pairs of symbols in the column [11].
SP - Score(X1#X2#...#Xk) = ∑ Cscore(X*1, i, X*2, i. . . X*k,i) (1) i = 0
Cscore(X*1, i, X*2, i. . . X*k,i) = ∑ Pscore(X*p,i , X*q,i) (2) 1≤p≤q≤k
3. 2. 1. 4. Crossover
In general, the crossover operator exchanges genes between two chromosomes to produce offsprings. The operation includes selection of a crossover point, genes from beginning to the selected point are copied from one parent, and the rest is copied from the second parent. The part of the chromosome within a crossover point consists of gap positions of a sequence. A chromosome may consist of several crossover points based on the no. of input sequences.
The developed tool provides two options for crossover operation (i) Single point crossover: any one of the crossover point is randomly selected to perform the operation. (ii) Double point crossover: any two crossover points are randomly selected to perform the operation.
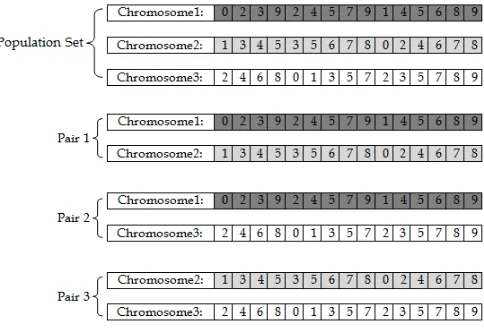
The chromosomes are selected for crossover operation by the following method:
First chromosome is crossover with the rest of the chromosomes in the population pool and the process con-tinues till the end as shown in Fig. 2.
Fig. 2: crossover operation
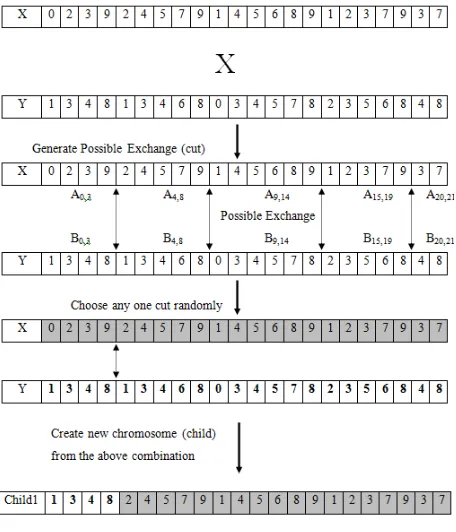
3. 2. 1. 5. Proposed Mutation Operator: X (Cross) – Shuffler
Mutation operator alters one or more gene values in a chromosome [23]. This is to prevent falling all solutions in population into a local optimum of solved problem [24].
The proposed operator is a hybrid of general crossover and new shuffler operator hence named as X-Shuffler operator.
In an X - shuffler process, the first step involves the crossover operation (single point/double point) as performed in the crossover section. This process is preferred before the mutation operator as it has the advantage of primitive random search, increases the possibility of getting best chromosomes.
As a second step either one or two mutation point is selected. The gaps positions in the blocks are re-placed with the shuffled gap positions to produce a new combination of number-string on two chromosomes resulting after the crossover operation. This is to avoid repeat and increase in number of gap position as an advantage. The algorithm terminates after the execution of given number of generations.
Fig. 3: X – Shuffler (Crossover part)
Fig. 4: X – Shuffler (Shuffler part)
3. 2. 2. PatternFinder
This module is used for identifying patterns in a set of aligned introns/exons sequences. The method is based on a matrix representation of binding site patterns. Each column of the matrix represents one of the four possible bases, each row represents one of the positions of the binding site and each element is determined by the frequency, the indicated base occurs at the indicated position [4].
number of sequences, while the time required increases only linearly with the number of sequences [4]. As an Example:
The pattern obtained is evaluated using the similarity percentage can be calculated by the formula: Similarity = (totally match column / maxLength) X 100
Where, maxLength is the alignment length which is calculated from N in chromosome representation. Therefore, the similarity of the pattern can be defined as:
Similarity = (5 / 7) X 100 = 71.42 %
The Table-2 shows, Similarity and TFS (%). TFS (%) is defined as TFS divided by the TFS with completely matched, i.e., if the length of pattern is 7 and the number of sequence 6, the TFS with completely matched is 7 x 6 = 42. Hence the TFS (%) = 1, if the pattern is complete matched in all sequences [2]. 4. Implementation
The web based tool iMAGA was written in JAVA and was implemented on a normal PC with an Intel Pentium IV processor rated at 3.0 GHz. The main memory is 1 GB (1024 megabytes). The operating sys-tem is Microsoft Windows XP SP3. Since it is web based, it contains both front end and back end parts. Front end was written in JSP, HTML and Java script. The back end was written in Servlet. In contrast, the Module I - iExtractor / iClassifier were written in BIOJAVA and Module II – iAligner / PatternFinder was written in JAVA. The server used for testing is Sun GlassFish Enterprise Server v3 and the software package used for developing package JCreator 4.50 Pro and for web application NetBeans IDE 6.8. Memory requirements are low, the main usage being to store the separate alignments in the population. For 20 sequences with an average alignment length of 200 and a population size of 100, ~2 Mb of memory is sufficient.
5. Experiment
The performance of the modified genetic algorithm is examined by analyzing the initial experimental results for processing time and the identified matches by taking intron as test data sets. This modified simple genetic algorithm is applied to four sets of intron sequences. These data sets differ with respect to their length, number, or similarity. The average and best results are obtained by continuously running our algorithm in fifty times. The match, mismatch, and space scores were +2, -2, and 0 respectively.
The maximum population size is 1000, generation is 5 and crossover type is 2. The X–Shuffler muta-tion operator, analogizes to the fragment based method, plays an important role in this modified simple genetic algorithm. When the alignment is made, the performance of genetic algorithm to solve MSA of introns is found to be good and the time complexity is given by
Time Complexity: (Number of sequences)(Average length of all sequences)
An example of possible alignments:
Number of sequences = 6
Average length of all sequences = 20
Then possible alignment is = 3656158440062976
Possible alignments of Intron sequence of Saccharomyces cerevisiae:
Average length of all sequences = 256
Then possible alignment is = 3.1902429234727776281076172435084e+1231
After the alignment process, PatternFinder – sub module identifies the pattern present in the set of aligned intron sequences. This displays the number of patterns found in the given intron sequences and their relevant information such as length, start and end position, graphical representation of intron sequences and their pattern.
6. Results and Discussion
iMAGA is a flexible tool for multiple sequence alignment and to find pattern. This can be seen by the ability of iMAGA to achieve optimal alignment scores and by the consistency of alignments with test cases of known intron sequences. The consistency of the iMAGA alignments with intron reference alignments is mainly a measure of the usefulness to find the pattern with the test data.
The alignment obtained from the widely used MSA tools (with default parameter settings) are compared with the alignment obtained from the developed iMAGA tool and the results are tabulated. Two standard reference datasets of DNA sequence alignments from BAliBASE [26] are used as input sequence.
Dataset Thy from smart_mdsa_100s version with 4 sequences.
Dataset RV20_BBS20001 from balibase_mdsa_all version with 16 sequences. Dataset RV11_BBS11022 from the balibase_mdsa_all version with 4 sequences.
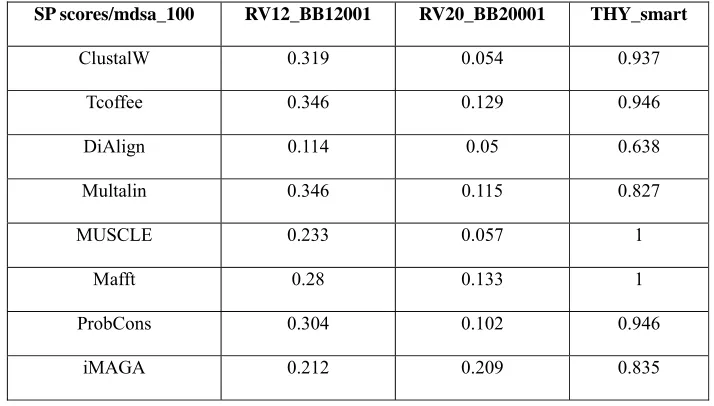
Table 1: SP score measurement using mdsa_100
SP scores/mdsa_100 RV12_BB12001 RV20_BB20001 THY_smart
ClustalW 0.319 0.054 0.937
Tcoffee 0.346 0.129 0.946
DiAlign 0.114 0.05 0.638
Multalin 0.346 0.115 0.827
MUSCLE 0.233 0.057 1
Mafft 0.28 0.133 1
ProbCons 0.304 0.102 0.946
iMAGA 0.212 0.209 0.835
The results obtained from the proposed iMAGA and other tools like Dialign, Mafft, ClustalW, T-Coffee, DiAlign, Multalin, MUSCLE, Mafft and ProbCons for multiple sequence alignment is analyzed using the SP score. It is observed that the alignment obtained from iMAGA is better than the compared existing tools.
Table 2: Comparison of predicted pattern for iMAGA with MEME
Tool
Pattern
Length
Predicted Pattern
Similarity
(%)
TFS (%)
(84% match)
Matched seqs#/Total
seqs# (90%)
iMAGA 7 GTA[TA]AG[CTA] 71.42 0.5 2/4
MEME 13
GTT[ACT][GT]T[AT]T[AT]C[CT][CG]
C
53.84 0.5 2/4
10 G[CT]TT[ACT][CG][AC][CT]TC 50 0.5 2/4
15
[GC]T[AT][AT]GT[AT][GC]T[AT][GC][
AT][TC][AT]T
33.33 0.25 1/4
Fig. 9: iAligner / PatternFinder Alignment Result
Fig. 10: iAligner / PatternFinder Pattern Result
7. Conclusion
iMAGA, a web based tool uses modified GA with newly designed mutation operator for alignment process where as some of the existing tools uses it for optimization of functioning algorithm. Moreover it also finds the pattern which may have significant role in deducing evolutionary relationship among different organisms. iMAGA is still fairly slow for large test cases (e.g. with >50 or so sequences). In future, it is desirable to use an optimization process with that of progressive approach in order to combine the speed of the former with the accuracy of the latter.
8. References
[1] Cédric Notredame and Desmond G. Higgins. (1996). SAGA: sequence Alignment by Genetic Algorithm, Nucleic Acids Research,
[2] Falcon F. M. Liu, Jeffrey J. P. Tsai, R. M. Chen, S. N. Chen, S. H. Shih. (2004). FMGA: Finding Motifs by Genetic Algorithm Fourth IEEE Symposium on Bioinformatics and Bioengineering (BIBE'04).
[3] Gelsema ES. (1995). Abductive reasoning in Bayesian belief networks using a gentic algorithm, Pattern Recog Lett, Vol. 16, pp
865-871.
[4] Gerald Z.Hertz, George W.Hartzell,III and Gary D.Stromo. (1990). Identification of consensus patterns in unaligned DNA sequences
known to be functionally related, Oxford Journals Bioinformatics, Vol. 6, No. 2, pp 81-92.
[5] Goldberg DE. (1989). Genetic Algorithms in Search, Optimization, and Machine Learning. Addison-Wesley, New York.
[6] Grate, L., and Ares, M. (2002). Searching Yeast Intron Data at the Areslab Website. (In Guide to Yeast Genetics and Molecular and
Cell Biology, Part B, C. Guthrie and G. Fink, eds) Methods Enz. Vol. 350, pp 380-392.
[7] Grefenstette JJ. (1986). Optimization of control parameters for genetic algorithms, IEEE Tans Syst Man Cybern Vol. 16, 122-128.
[8] J.D. Thompson, J.e. Thierry, O. Poch. (2003). RASCAL: rapid scanning and correction of multiple sequence alignments,
Bioinformatics, Vol. 19, No. 9.
[9] Javid Taheri and Albert Y Zomaya. (2009). RBT-GA: A Novel Metaheuristic for Solving The Multiple Sequence Alignment Problem,
BMC Genomics, 10(Suppl 1):S10.
[10] Jerry Bergman. (2001). The Function of Introns: From Junk DNA to Designed DNA. Perspectives on Science and Christian Faith Vol.
53, No. 3.
[11] Jorng-Tzong Horng, Li-Cheng Wu, Ching-Mei Lin, Bing-He Yang. (2005). A Genetic Algorithm For Multiple Sequence Alignment,
Soft Computing-A Fusion of Foundations, Methodologies and Applications, Vol. 9, Issue 6, pp 407 – 420.
[12] Koji Tajima. (1993). Multiple Sequence Alignment using Parallel Genetic Algorithms, Genome Informatics, Vol. 4, pp 183-187.
[13] Kosmas Karadimitriou and Donald H. Kraft. (1996). Genetic Algorithms And The Multiple Sequence Alignment Problem In Biology,
Proceedings of the Second Annual Molecular Biology and Biotechnology Conference.
[14] M. F. Omar, R. A. Salam, R. Abdullah, N. A. Rashid. (2005). Multiple Sequence Alignment Using Optimization Algorithms,
International Journal of Computational Intelligence, 1; 2.
[15] Masamichi Isokawa Masato Wayama Toshio Shimizu. (1996). Multiple Sequence Alignment Using a Genetic Algorithm, Genome
Informatics, Vol. 7, pp 176-177.
[16] S. Marsili Libelli, P. Alba. (2000). Adaptive mutation in genetic algorithms, Soft Computing, Vol. 4, pp 76 – 80.
[17] Sendhoff B, Kreutz M, Seelen W. (1997). Causality and the analysis of local search in evolutionary algorithms, internal report 97-16, Institüt für Neuroinformatik, - Ruhr-Universitat Bochum.
[18] Setubal J, Meidanis J. (1997). Sequence comparison and database search. Introduction to Computational Molecular Biology, PWS, pp.
47–103.
[19] T. Jiang, L. Wang. (1994). On the complexity of multiple sequence alignment, J. Comput. BioI. Vol. 1, pp 337-378.
[20] Wayama M, Takahashi K, Shimizu T. (1995). An approach to amino acid sequence alignment using a genetic algorithm. Genome
Informatics, Vol. 6, pp 122–123.
[21] Whitley D. (1994). A genetic algorithm tutorial. Statistics and Computing, Vol. 4, pp 65–85.
[22] Yang Chen, Jinglu Hu, Member, IEEE, Kotaro Hirasawa, Member, IEEE, Songnian Yu. (2008). Multiple Sequence Alignment Based
on Genetic Algorithms with Reserve Selection ICNSC, pp 1511-1516.
[23] O. Roeva, A. Shannon. (4th July 2008). A Generalized Net Model of a Mutation Operator for the Breeder Genetic Algorithm, Ninth Int. Workshop on GNs, Sofia, pp 59-63.
[24] Rini Mahajan, Amit Saxena, Baljit Singh Khehra. (2010). A Genetic Approach to Standard Cell placement using Various Genetic
Operators, International Journal of Computer Applications, Vol. 1-No. 9, pp 0975-8887.
[25] http;//www.rae.org/introns.html, The Functions of Introns: From Junk DNA to Designed DNA by Jerry Bergman
[26] Hyrum D.Carroll et.al (2007): “DNA reference alignment benchmarks based on tertiary structure of encoded proteins”,