inorganic papers
Acta Cryst.(2007). E63, i105–i107 doi:10.1107/S1600536807010483 Wanget al. CdHPO
3
i105
Acta Crystallographica Section E Structure Reports Online
ISSN 1600-5368
CdHPO
3with a channel structure
Li Wang, Tian-You Song, Jia-Ning Xu and Su-Hua Shi*
College of Chemistry, Jilin University, Jiefang Road 2519, Changchun, People’s Republic of China
Correspondence e-mail: [email protected]
Key indicators
Single-crystal X-ray study
T= 293 K
Mean(P–O) = 0.003 A˚
Rfactor = 0.027
wRfactor = 0.067
Data-to-parameter ratio = 13.9
For details of how these key indicators were automatically derived from the article, see http://journals.iucr.org/e.
Received 7 December 2006 Accepted 5 March 2007
#2007 International Union of Crystallography
All rights reserved
Cadmium phosphite, CdHPO3, was obtained by mild
hydro-thermal synthesis. The construction of the three-dimensional framework structure may be viewed as an assembly of distorted CdO6 octahedra and HPO3 tetrahedra which leads
to the formation of channels along [001]. The crystal structure is isotypic with CdSO3-III and is a new example of the
stereochemical equivalence of the P-bonded H atom in HPO32 and the electron lone pair in XO32 oxoanions (X
= SIVor SeIV).
Comment
Recently, compounds containing the tetrahedral phosphite group, HPO3
2
[image:1.610.230.437.481.692.2], have been investigated as alternative candi-dates for a possible replacement of phosphates in their various applications. Like the corresponding phosphates, transition metal phosphites exhibit a multi-faceted crystal chemistry with the formation of one-dimensional chain structures, two-dimensional layer structures or three-two-dimensional framework structures (Shiehet al., 1990; Zakharovaet al., 1994; Sghyaret al., 1991; Sapina et al., 1989; Liu et al., 2005). However, the number of structurally well characterized transition metal phosphites is much smaller than that of the corresponding phosphates. In the course of studies of the formation of novel transition metal phosphites, we have successfully employed
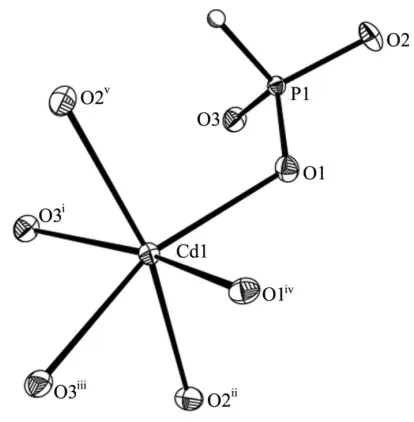
Figure 1
The CdO6octahedron and an attached PO3H tetrahedron in the structure
of (I), with displacement ellipsoids drawn at the 50% probability level. The H atom is shown as a sphere of arbitrary radius. [Symmetry codes: (i) 1x,y,z(ii)2
3y, 2 3+xy,
1
3z(iii) 1
3+xy, 2 3+x,
1 3z(iv) 1
3+y, 2 3x+y,
2
hydrothermal methods and have prepared anhydrous cadmium phosphite, CdHPO3, (I). Its structure is isotypic with
CdSO3-III (Engelen et al., 1987), and therefore (I) can be
viewed as another example of the stereochemical equivalence of the P-bonded H atom in HPO3
2
and the electron lone pair in XO3
2
oxoanions (X = SIV or SeIV) (Corbridge, 1956; Gattow, 1958; Weisset al., 1966; Handlovicˇ, 1969; Andersenet al., 1984; Engelenet al., 1987; Boldtet al., 2000).
In the asymmetric unit of compound (I) there are one Cd, one P, one H and three O sites (Fig. 1). The Cd atom has a distorted octahedral coordination by six O atoms, with Cd—O bond lengths ranging from 2.233 (3) to 2.353 (3) A˚ (Table 1). The P atom is in a distorted tetrahedral coordination by three O atoms, with P—O bond lengths between 1.521 (3) and 1.529 (3) A˚ , and by one H atom, with a bond length of 1.38 (5) A˚ , in good agreement with other phosphite structures reported previously (Bonaviaet al., 1995). The existence of a P—H bond is verified by IR spectroscopy, which reveals a characteristic strong absorption at 2402 cm1(Shiet al., 2004). The construction of the three-dimensional framework structure in compound (I) may be viewed as an assembly of CdO6 octahedra and distorted HPO3 tetrahedra sharing
corners and edges. The CdO6octahedra share edges with O2
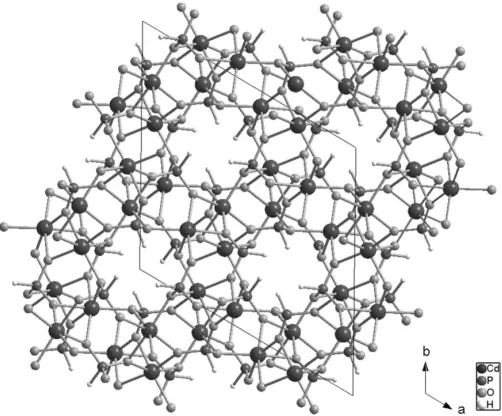
and O3 as bridging atoms, which leads to the formation of pseudo-helical chains. Adjacent chains are linked by HPO3
tetrahedra to construct a three-dimensional framework with channels along [001], as shown in Fig. 2. The free space within the channels is slightly reduced because the H atoms of the phosphite tetrahedra protrude towards the centre of the channels.
Experimental
Compound (I) was prepared from a reaction mixture consisting of CdCl22.5H2O, H3PO3, ethylenediamine and water in a molar ratio of
1:12:2:444. A typical reaction batch consisted of CdCl22.5H2O
(0.228 g), H3PO3(1.0 g), ethylenediamine (0.10 ml) and water (8 ml).
The mixture was stirred for 30 min at room temperature, transferred to a 23 ml PTFE-lined stainless steel autoclave with a filling capacity of 50% and heated at 443 K for 5 d under autogenous pressure. Fine colourless crystals of (I) with rod or block-like habit were filtered off, washed with water and dried at room temperature (yield 60% based on Cd).
Crystal data
CdHPO3 Mr= 192.39
Trigonal,R3 a= 13.509 (2) A˚ c= 8.379 (2) A˚ V= 1324.3 (4) A˚3
Z= 18
MoKradiation
= 7.73 mm1 T= 293 (2) K 0.200.200.18 mm
Data collection
Bruker SMART APEX CCD area-detector diffractometer Absorption correction: none 2018 measured reflections
679 independent reflections 660 reflections withI> 2(I) Rint= 0.024
Refinement
R[F2> 2(F2)] = 0.027 wR(F2) = 0.067 S= 1.03 679 reflections
49 parameters
All H-atom parameters refined
max= 0.92 e A˚ 3
min=3.51 e A˚ 3
Table 1
Selected geometric parameters (A˚ ,).
Cd1—O1 2.233 (3)
Cd1—O3i 2.275 (3)
Cd1—O2ii 2.322 (3) Cd1—O3iii 2.325 (3) Cd1—O1iv 2.333 (3) Cd1—O2v 2.353 (3)
P1—O3 1.521 (3)
P1—O2 1.523 (3)
P1—O1 1.529 (3)
P1—H1 1.38 (5)
O3—P1—O2 111.91 (16)
O3—P1—O1 111.73 (16)
O2—P1—O1 111.22 (17)
O3—P1—H1 109 (2)
O2—P1—H1 107 (2)
O1—P1—H1 105 (2)
Symmetry codes: (i) xþ1;y;z; (ii) yþ2 3;xy
2 3;zþ
1 3; (iii) xy1
3;x23;zþ31; (iv)yþ13;xþyþ23;zþ23; (v)xþyþ1;xþ1;z.
The H atom was located in a difference Fourier map and refined freely. The deepest hole in the final Fourier map is 0.02 A˚ away from Cd.
Data collection:SMART(Bruker, 2003); cell refinement: SAINT-Plus(Bruker, 2003); data reduction:SAINT-Plus; program(s) used to solve structure: SHELXS97(Sheldrick, 1997); program(s) used to refine structure:SHELXL97(Sheldrick, 1997); molecular graphics:
SHELXTL (Bruker, 2003) and DIAMOND (Brandenburg, 2005);
software used to prepare material for publication:SHELXL97.
This work was supported by the State Basic Research Project (grant No. G2000077507) and the National Science Foundation of China (grant Nos. 29873017 and 20101004).
References
Andersen, L., Lindqvist, O. & Moret, J. (1984).Acta Cryst.C40, 586–589. Boldt, K., Engelen, B., Pantho¨fer, M. & Unterweide, K. (2000).Eur. J. Inorg.
Chem.pp. 2071–2075.
inorganic papers
i106
Wanget al. CdHPO [image:2.610.46.297.71.279.2]3 Acta Cryst.(2007). E63, i105–i107
Figure 2
Bonavia, G., Debord, J., Haushalter, R. C., Rose, D. & Zubieta, J. (1995). Chem. Mater.7, 1995–1998.
Brandenburg, K. (2005).DIAMOND. Release 3.1. Crystal Impact GbR, Bonn, Germany.
Bruker (2003).SMART,SAINT,SHELXTLandSADABS. Bruker AXS Inc., Madison, Wisconsin, USA.
Corbridge, D. E. C. (1956).Acta Cryst.9, 991–994.
Engelen, B., Buchmeier, W. & Lutz, H. D. (1987).Z. Naturforsch. Teil B,42, 37–41.
Gattow, G. (1958).Acta Cryst.11, 377–383. Handlovicˇ, M. (1969).Acta Cryst.B25, 227–231.
Liu, W., Chen, H. H., Yang, X. X. & Zhao, J. T. (2005).Eur. J. Inorg. Chem.pp. 946–951.
Sapina, F., Gomez-Romero, P., Marcos, M. D., Amoros, P., Ibanez, R., Beltran, D., Navarro, R., Rillo, C. & Lera, F. (1989).Eur. J. Solid State Inorg. Chem.
26, 603–617.
Sghyar, M., Durand, J., Cot, L. & Rafiq, M. (1991).Acta Cryst.C47, 2515–2517. Sheldrick, G. M. (1997). SHELXS97 and SHELXL97. University of
Go¨ttingen, Germany.
Shieh, M., Martin, K. J., Squattrito, P. J. & Clearfield, A. (1990).Inorg. Chem.
29, 958–963.
Shi, S. H., Qian, W., Li, G. H., Wang, L., Yuan, H. M., Xu, J. N., Zhu, G. S., Song, T. Y. & Qiu, S. L. (2004).J. Solid State Chem.177, 3038–3044. Weiss, R., Wendling, J.-P. & Grandjean, D. (1966).Acta Cryst.20, 563–566. Zakharova, B. S., Ilyukhin, A. B. & Chudinova, N. N. (1994).Zh. Neorg. Khim.
39, 1443–1445. (In Russian.)
inorganic papers
Acta Cryst.(2007). E63, i105–i107 Wanget al. CdHPO
supporting information
sup-1 Acta Cryst. (2007). E63, i105–i107
supporting information
Acta Cryst. (2007). E63, i105–i107 [https://doi.org/10.1107/S1600536807010483]
CdHPO
3with a channel structure
Li Wang, Tian-You Song, Jia-Ning Xu and Su-Hua Shi
Cadmium phosphate(III)
Crystal data CdHPO3 Mr = 192.39 Hexagonal, R3 Hall symbol: -R 3 a = 13.509 (2) Å c = 8.379 (2) Å V = 1324.3 (4) Å3 Z = 18
F(000) = 1584
Dx = 4.342 Mg m−3
Mo Kα radiation, λ = 0.71073 Å Cell parameters from 2109 reflections θ = 3.0–27.3°
µ = 7.73 mm−1 T = 293 K Block, colourless 0.20 × 0.20 × 0.18 mm
Data collection
Bruker SMART APEX CCD area-detector diffractometer
Radiation source: fine-focus sealed tube Graphite monochromator
ω scans
2018 measured reflections 679 independent reflections
660 reflections with I > 2σ(I) Rint = 0.024
θmax = 27.4°, θmin = 3.0° h = −10→17
k = −17→17 l = −9→10
Refinement Refinement on F2 Least-squares matrix: full R[F2 > 2σ(F2)] = 0.027 wR(F2) = 0.067 S = 1.03 679 reflections 49 parameters 0 restraints
Primary atom site location: structure-invariant direct methods
Secondary atom site location: difference Fourier map
Hydrogen site location: difference Fourier map All H-atom parameters refined
w = 1/[σ2(F
o2) + (0.055P)2 + 5.8207P] where P = (Fo2 + 2Fc2)/3
(Δ/σ)max = 0.012 Δρmax = 0.92 e Å−3 Δρmin = −3.51 e Å−3
Special details
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes.
supporting information
sup-2 Acta Cryst. (2007). E63, i105–i107
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
x y z Uiso*/Ueq
Cd1 0.44451 (2) 0.06208 (2) 0.23950 (3) 0.00916 (16)
P1 0.68801 (8) 0.16748 (8) 0.04639 (11) 0.0072 (2)
O1 0.6343 (2) 0.1537 (2) 0.2114 (3) 0.0114 (5)
O3 0.6532 (2) 0.0525 (2) −0.0300 (3) 0.0131 (6)
O2 0.8174 (2) 0.2440 (2) 0.0533 (3) 0.0135 (6)
H1 0.646 (5) 0.224 (5) −0.043 (6) 0.016*
Atomic displacement parameters (Å2)
U11 U22 U33 U12 U13 U23
Cd1 0.00783 (19) 0.00993 (19) 0.0091 (2) 0.00400 (12) −0.00008 (8) 0.00032 (8)
P1 0.0065 (4) 0.0077 (4) 0.0078 (4) 0.0039 (3) −0.0001 (3) −0.0001 (3)
O1 0.0094 (12) 0.0139 (13) 0.0111 (12) 0.0059 (11) 0.0005 (9) −0.0013 (10)
O3 0.0154 (13) 0.0108 (13) 0.0128 (13) 0.0064 (11) −0.0026 (11) −0.0023 (10)
O2 0.0076 (13) 0.0126 (13) 0.0164 (13) 0.0022 (10) 0.0018 (10) −0.0040 (11)
Geometric parameters (Å, º)
Cd1—O1 2.233 (3) P1—O1 1.529 (3)
Cd1—O3i 2.275 (3) P1—H1 1.38 (5)
Cd1—O2ii 2.322 (3) O1—Cd1vi 2.333 (3)
Cd1—O3iii 2.325 (3) O3—Cd1i 2.275 (3)
Cd1—O1iv 2.333 (3) O3—Cd1vii 2.324 (3)
Cd1—O2v 2.353 (3) O2—Cd1viii 2.322 (3)
P1—O3 1.521 (3) O2—Cd1ix 2.354 (3)
P1—O2 1.523 (3)
O1—Cd1—O3i 114.21 (10) O3—P1—O2 111.91 (16)
O1—Cd1—O2ii 98.53 (10) O3—P1—O1 111.73 (16)
O3i—Cd1—O2ii 91.52 (10) O2—P1—O1 111.22 (17)
O1—Cd1—O3iii 159.24 (10) O3—P1—H1 109 (2)
O3i—Cd1—O3iii 85.76 (3) O2—P1—H1 107 (2)
O2ii—Cd1—O3iii 74.24 (10) O1—P1—H1 105 (2)
O1—Cd1—O1iv 83.21 (13) P1—O1—Cd1 120.41 (16)
O3i—Cd1—O1iv 159.60 (10) P1—O1—Cd1vi 118.53 (15)
O2ii—Cd1—O1iv 96.41 (9) Cd1—O1—Cd1vi 109.52 (11)
O3iii—Cd1—O1iv 78.41 (10) P1—O3—Cd1i 142.90 (17)
O1—Cd1—O2v 91.92 (10) P1—O3—Cd1vii 114.33 (15)
O3i—Cd1—O2v 74.57 (10) Cd1i—O3—Cd1vii 102.68 (11)
O2ii—Cd1—O2v 165.19 (10) P1—O2—Cd1viii 126.47 (16)
O3iii—Cd1—O2v 99.24 (10) P1—O2—Cd1ix 131.89 (16)
O1iv—Cd1—O2v 95.21 (10) Cd1viii—O2—Cd1ix 100.37 (10)
Symmetry codes: (i) −x+1, −y, −z; (ii) −y+2/3, x−y−2/3, z+1/3; (iii) x−y−1/3, x−2/3, −z+1/3; (iv) y+1/3, −x+y+2/3, −z+2/3; (v) −x+y+1, −x+1, z; (vi)