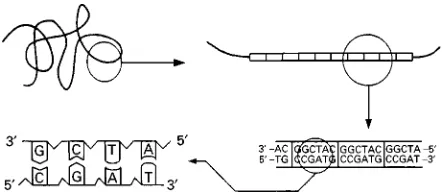
Figure 1 Schematic description at four increasing levels of magnification for DNA and VNTRs.
DEOXYRIBONUCLEIC ACID PROFILING
Overview
R. Coquoz, Institut de Police Scientifique et de Criminologie, Lausanne, Switzerland
Copyright^ 2000 Academic Press
This article is reproduced fromEncyclopedia of Analyti-cal Science, Copyright^ 1995 Academic Press
Introduction
The forensic biologist has traditionally used a large set of markers including blood group antigens and serum proteins to establish links between evidence and comparison samples. These markers are poly-morphic and genetically determined. In 1985, a new technique was introduced which opened the door to the forensic use of deoxyribonucleic acid (DNA) technology: its inventor coined the term ‘DNAR nger-printing’. Since then, DNA typing in this and many other forms hasSourished and the forensic biologist now has access to a large variety of powerful techniques.
DNA and its Polymorphism
DNA is an extremely large linear molecule which stores genetic information in the form of a sequence of its constituent elements: the nucleotides. The DNA molecule is made up of two complementary chains or strands of nucleotides (Figure 1). There are four different nucleotides (symbolized by the letters A, C, T, G) whose chemical afRnity determines the complementarity of the nucleotide sequences of the two strands of the DNA molecule. Nucleo-tides A and T, and G and C always face each other; as a consequence, the length of DNA fragments is usually expressed in ‘base pair’ (bp) units. This chem-ical afRnity allows single strands of complement-ary DNA to hybridize to each other. Nucleotide chains are oriented, having so called 5and 3 ends, and the two strands of DNA have an opposite ori-entation. The DNA synthesis occurs in the direction 5P3.
The polymorphism within DNA resides partly in the existence of minor nucleotide sequence variations among different individuals at various locations (loci) in the DNA molecule, which has its counterpart in the polymorphism of the amino acid sequence of the proteins. The gene variants (the alleles) differ
in their sequence. However, a more extensive and DNA-speciRc polymorphism exists in the noncoding DNA: the tandemly repeated sequences. These poly-morphic loci are DNA fragments, made up of a nu-cleotide sequence (the repeat unit), tandemly repeated like a series of identical beads on a necklace (Fig-ure 1). The repeat unit can be from two to more than 100 nucleotides long. The polymorphism comes from the extreme variation in the number of repeats. The alleles differ then by their size (length polymor-phism). This kind of repetitive sequence has been called minisatellites or also variable number tandem repeat (VNTR). There is a huge reservoir of polymor-phism in these sequences and it is their potential use which has stimulated the development of the various DNA typing procedures.
DNA Typing Procedures
Any biological material containing DNA may be use-ful, including blood, sperm, saliva, hair, autopsy tis-sue, bone. All DNA typing procedures involve a DNA isolation step, which allows the elimination of sub-stances such as proteins which can interfere in the sometimes delicate enzymatic steps involved in any DNA typing method.
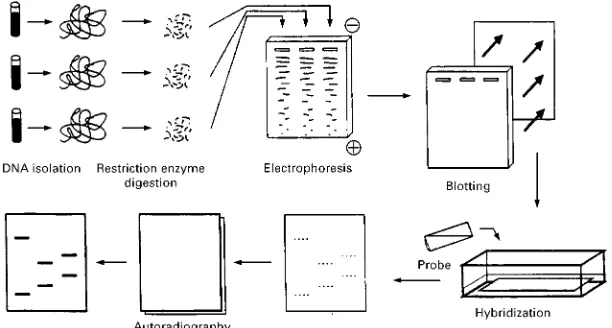
Figure 2 Schematic description of the main steps in RFLP typing. Restriction Fragment Length Polymorphism (RFLP) Typing
The isolated DNA is reproducively cut into fragments by the action of a sequence speciRc DNA cutting enzyme, a restriction enzyme (hence RFLP). The frag-ments, which can be more than 20 000 bp long, are then separated according to their sizes by elec-trophoresis on agarose gel. The DNA fragments are transferred to a nylon membrane to which they will remain attached but are still accessible for hybridiza-tion with a probe. This membrane will provide a rep-lica of the electrophoresis product. Labelled pieces of DNA having a sequence complementary to the repeti-tive sequence to be detected (the probe) are added to the nylon membrane and hybridize to the appropriate DNA fragments. These can Rnally be detected by autoradiography with aRlm sensitive to the labelled probe (Figure 2). The DNA fragments to be detected will appear on theRlm as dark bands (Figures 2and 3). Their position will be a measure of their size, which is itself proportional to the number of repeti-tive elements in the VNTR. A comparison with DNA fragments of known sizes allows an accurate size determination. ThisRrst probe can be stripped from the membrane allowing for reprobing with a second probe, etc. There is a large choice of VNTRs available for RFLP typing (Table 1). It is the same for restric-tion enzymes: but a few have found wider use because of their robustness and their ability to cut DNA into small fragments. These enzymes are Hae III, Hinf I, Pst I, Alu I and Pvu II, theRrst two being the most used. The labelling of the probe is traditionally done through the use of 32P-labelled nucleotides.
However, probes coupled to enzymes catalysing the production of a chemiluminescent substance allow detection limits as low as those obtained by radioac-tive means.
The probe can be designed so as to detect a single VNTR locus; it is then a single locus probe (SLP). If the individual has inherited DNA from his parents with a different number of repeats at the ana-lysed VNTR, the result is in the form of two bands (heterozygote); otherwise, there will be a single band (homozygote). But since the repeat units of dif-ferent VNTRs often have sequence similarities to each other, it is possible to detect a whole family of VNTRs at the same time provided a probe is used under conditions allowing hybridization with par-tially complementary DNA fragments. In this case we speak of a multilocus probe (MLP) and the result appears in the form of a set of bands with a very individual pattern similar to a bar code.
Polymerase Chain Reaction (PCR)
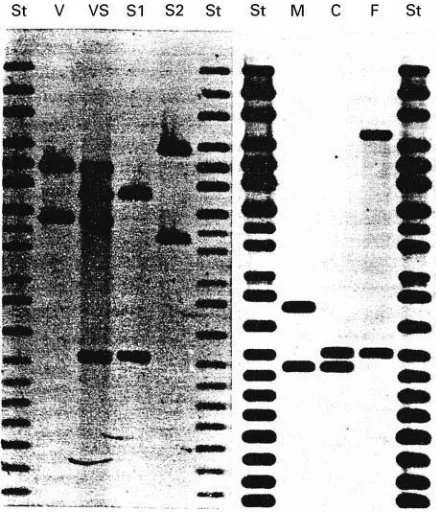
Figure 3 Typical examples of RFLP typing of forensic cases. On the left: sexual assault case; St, size markers, V, victim, VS, vaginal swab (containing material from the victim and her assail-ant), S1, suspect 1, S2, suspect 2. On the right: paternity test with M, mother, C, child, F, putative father.
Table 1 Selection of some of the main DNA loci analysed by the various DNA typing methods Locus Corresponding
probe
Chromosome location
Repeat unit length (bp)
Typing method used Heterozygosityb(%)
* 33.6 1a 37a multilocus RFLP *
D1S 7 MS1 1 9 monolocus RFLP '99
D7S21 MS31 7 20 monolocus RFLP 98
D2S44 YNH24 2 31 monolocus RFLP 94
D10S28 TBQ7 10 33 monolocus RFLP 97
HLA-DQ * 6 * PCR#dot blotting 79
D1S80 MCT 118 1 16 AMP-FLP 80
D17S5 YNZ22 17 70 AMP-FLP 82
APOC2 Mdf 5 19 2 STR 80
HUMTH01 * 11 4 STR 80
aThese characteristics refer to the VNTR used as the multilocus probe.
bHeterozygosity is calculated here as the percentage of apparent heterozygotes in the population (Caucasian in this case). makes the whole analytical process convenient and
rapid.
PCR and Dot+Blot Analysis
Many methodologies have been designed which make use of the main principle of a PCR. In forensic science, PCR wasRrst used to analyse sequence poly-morphisms such as the polymorphism of the HLA-DQ locus. In that case, the alleles, which differ only by a few nucleotide changes, are identiRed after use of a PCR through hybridization with
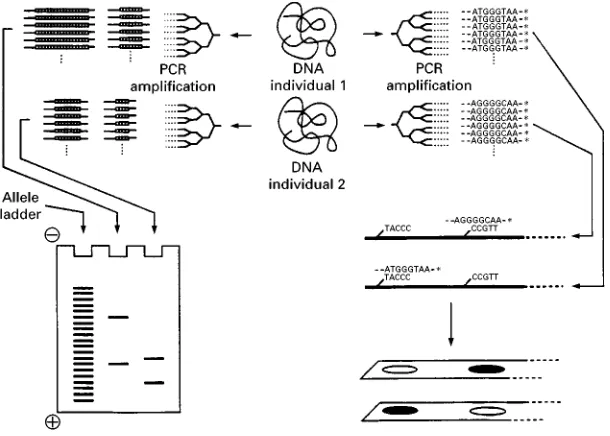
allele-speci-Rc probes, using a reverse dot}blot format (Figure 4): the various probes are attached to a membrane strip, arranged as a series of invisible dots to which the ampliRcation products can hybridize depending upon thier sequence. Through the use of labelled nucleo-tides during the PCR, the genotype of the sample can be read through the appearance or not of coloured dots on the strip, at the position of each probe (Fig-ure 4). In a way, the format is not very different from that of the strips for urine analysis so widely used in clinical medicine.
Ampli\cation Fragment Length Polymorphism (AMP-FLP) Typing
Another use of a PCR has been the ampliRcation of VNTRs to analyse their length polymorphism. How-ever, the quite poor performance of PCRs in the ampliRcation of large DNA fragments, such as those of the VNTR loci analysed through RFLP typing, has led to the search for smaller VNTRs (Table 1). By designing primers located on each side of a VNTR, it can be ampliRed by a PCR and the ampliRed frag-ments can be analysed by electrophoresis on agarose or polyacrylamide gels and compared to standards (Figure 4). While RFLP typing required the use of probes to detect speciRc VNTR-DNA fragments among thousands of other fragments, the ampliR ca-tion process here allows the use of nonspeciRc detec-tion methods (ethidium bromide staining, silver stain-ing) to make the bands visible. This kind of PCR-analysed VNTR polymorphism has been called
ampli-Rcation fragment length polymorphism (AMP-FLP).
Short Tandem Repeats (STR) Typing
[image:3.568.52.517.552.689.2]Figure 4 A PCR amplification for AMP-FLP typing (left) and reverse dotIblot analysis (right).
or short tandem repeats (STRs) and they are probably the richest class of polymorphic loci available. Their ampliRcation conditions are simple and the size range of their alleles is usually narrow. They are then well suited to multiplexing: it is indeed easy to choose microsatellite loci and adequate primers so as to ob-tain ampliRed DNA fragments situated in well separ-ated size ranges for each locus. Consequently a whole group of STRs can be ampliRed at the same time and analysed on the same gel. The small size of their repeat unit, however, is also a disadvantage. The various alleles only differ in length by two, three or four nucleotides. Such a separation power is only possible through the more complex technology of sequencing gel electrophoresis. However, the se-quencing technology is undergoing constant improve-ment due to the challenge of the human genome sequencing project, and STR typing has beneRted from the developments in that Reld such as S uor-escent labelling and automatic sequencing.
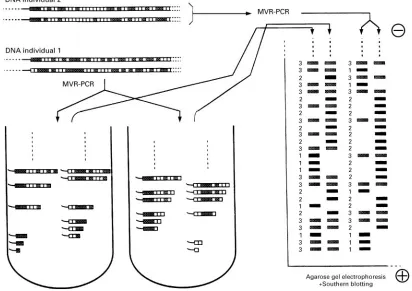
Minisatellite Variant Repeat (MVR)-PCR
The polymerase chain reaction has made possible yet another powerful DNA typing method: MVR-PCR. This has been designed to reveal a polymorphism consisting of variations in the sequence of the repeat unit within some VNTRs. Instead of being a pure repetition of one repeat unit, some VNTRs are com-posed of combinations of two or more almost identi-cal repeat units. It is easy to identi-calculate that even with only two repeat variants a and b, the number of possible combinations of repeats is huge. The se-quence of repeats is revealed through a process some-hat similar to the Sanger DNA sequencing method
Figure 5 Schematic description of MVR-PCR.
result is literally read by itself, and different samples are simply compared at the level of their codes.
Other Methods
Other DNA typing methods have been developed or proposed, and still others are yet to appear. Two methods are worth mentioning. One is mitochondrial DNA (mtDNA) sequence analysis. A DNA sequence analysis after PCR ampliRcation is a natural way to examine DNA polymorphism. However, because it is unable to reveal more than a few hundred base pairs at a time, and because of its complexity, it is not as attractive as the above-mentioned methods. How-ever, it has been a precious tool in the analysis of mitochondrial DNA, which is a small piece of DNA, maternally inherited, present in all mitochondria. Part of it (the D-loop) contains a highly polymorphic sequence and its sequence determination is therefore very informative. Owing to the presence of more than 1000 copies in each cell. mtDNA analysis is very sensitive. Moreover, hair shafts, which do not contain nuclear DNA, are rich in mtDNA and are therefore amenable to genetic analysis.
The second method is polymorphic sequence-tagged sites (pSTS) analysis. This is a method de-signed to reveal single base pair changes. After a PCR ampliRcation of the DNA fragment containing the
polymorphic base pair, a ligation assay is performed using an antigen-labelled primer and one of two biotin-labelled primers each being speciRc for one of the two possible alleles. In the presence of the cor-responding sample allele, the biotin-labelled primer is ligated to the antigen-labelled primer. After capture of the product in microtitre plates coated with strep-tavidin (a binding protein for biotin), the presence of the antigen can be detected using a traditional immunoenzymatic method (ELISA procedure). The combined analysis of more than 15 such two-allele sites can reveal genotypes with frequencies below 10\6. The most interesting features of this method
are its technical simplicity and its straightforward interpretation (positive or negative readout) which make it particularly amenable to automation. It could reach its highest performance if all the loci could be analysed simultaneously within the same tube.
Applications
sequential analysis of more than 20 polymorphic sys-tems, each requiring its own analytical process. The improvement is particularly evident in semen analysis where the number of available polymorphic systems was extremely low. In RFLP typing, the analysed VNTR loci are so polymorphic that most of the possible genotypes have frequencies below 1%. It is then easy to calculate that the combined analysis of four such loci reveals genotypes with frequencies of the order of 10\8. Both AMP-FLP typing and STR
typing use smaller, less polymorphic and therefore less informative VNTRs, but a similarly high in-formation yield can be reached by increasing the number of loci analysed.
Interpretation
Forensic identiRcation is achieved through the dem-onstration of matching biological characteristics be-tween evidence and comparison material. The match-ing is straightforward when discrete and well-identiR -able alleles are analysed such as in the various methods derived from a PCR. However, in RFLP typing, a match is not always easy to establish. First there is the intrinsic difRculty of having to deal with alleles which cannot each be differentiated by elec-trophoresis. Indeed, because of the size range of the DNA fragments, the separation power of elec-trophoresis is insufRcient to separate alleles differing in length by only one repeat unit. Two samples hav-ing bands at an identical position may in fact possess different alleles. Secondly, there is the possibility that a DNA sample originating from the same person as the comparison sample contains interfering molecu-les, leading to a different electrophoretic behaviour and shifted band positions. This bandshifting can only be demonstrated and evaluated through the use of proper controls (internal standards).
The next step in the interpretation is the evaluation of the power of the evidence. It is usually expressed as a ratio of the probability of the evidence under the hypothesis that the evidential material has the same source as the comparison sample and the probability of the evidence under the hypothesis that somebody else is the source of the evidence material. This ratio is normally inversely proportional to the frequency of the genetic characteristics detected in the adequate reference populaton. The determination of these fre-quencies is a difRcult task. The extreme variability of the VNTR loci makes the gathering of genotype quency data unrealistic. Statistically trustworthy fre-quencies would require the analysis of a huge number of individuals. It is thus necessary to rely on allele frequencies to calculate the genotype frequencies by multiplication. The frequency of genotype A1A2 at
locusAis the product 2a1a2(witha1anda2being the
respective allele frequencies). The frequency of the combined genotypes at four loci is then 2a1a2;
2b1b2;2c1c2;2d1d2. However, these multiplications
can only be done under the assumption of statistical independence of the multiplied factors. This trans-lates into certain assumptions related to the genetic independence of the analysed VNTR loci and to the genetic structure of the populations (Hardy-Weinberg equilibrium).
Controversy has arisen over the possibility of the existence of genetic substructuring within the popula-tions. Some geneticists expressed concern that the frequency of a genotypeA1A2might have an apparent
value using the general population database, but that the true frequency might be much higher owing to the high frequency of these speciRc alleles in a genetically distinct population subgroup. At the extreme, if allelesA1A2only appear in this subpopulation where
they have a frequency of 50% each, the detection of allele A1 in a sample would make the detection of
alleleA2almost certain. Because of such effects,
com-bined genotype frequencies could be underestimated by several orders of magnitude. The accumulating data suggest, however, that there is not much reason to worry about such effects. The VNTR loci are extremely polymorphic in every population studied and, for any given genotype constellation, there is an extremely low probability of having substantial fre-quency differences when calculating with one popula-tion data or another. Moreover, within the major races, the frequency distributions are very similar between different populations. AndRnally, forensic laboratories use very conservative approaches which always favour the accused at each interpretation step.
Paternity Testing
The identiRcation power of DNA typing has naturally been used in paternity testing (Figure 3). But here the question relates to the transmission of genetic mater-ial from one generation to the other. Mutations which can cause apparent exclusions of true fathers then have to be taken into account. The polymor-phism of the VNTRs is linked to an exceptionally high mutation rate. This mutation rate can be below 10\4to more than 0.05 mutations, per meiosis for the
most polymorphic loci such asD1S7. This has to be compared to the mutation rate of coding DNA se-quences which is estimated to be around 10\5 for
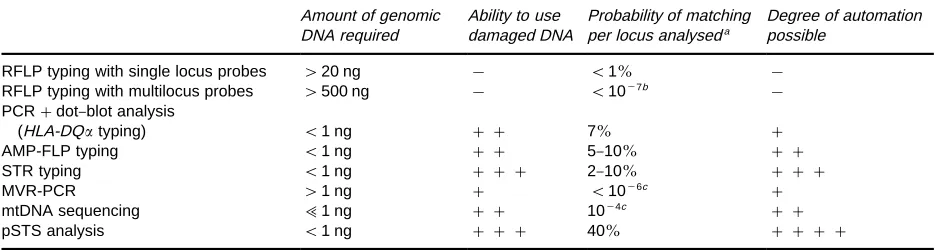
Table 2 Main characteristics of the various techniques available for DNA typing Amount of genomic
DNA required
Ability to use damaged DNA
Probability of matching per locus analyseda
Degree of automation possible
RFLP typing with single locus probes '20 ng ! (1% !
RFLP typing with multilocus probes '500 ng ! (10\7b !
PCR#dot}blot analysis
(HLA-DQtyping) (1 ng ## 7% #
AMP-FLP typing (1 ng ## 5}10% ##
STR typing (1 ng ### 2}10% ###
MVR-PCR '1 ng # (10\6c #
mtDNA sequencing ;1 ng ## 10\4c ##
pSTS analysis (1 ng ### 40% ####
aThe probability of matching (Pm) is the probability that two individuals taken at random have the same genotype. Pm"n i"1P
2 i wherePiis the frequency of the genotype for the analysed locus.
bPm is that for the whole set of loci detected by this probe.
cThese numbers are estimates based on limited population samples. Practical Use
In the real world of forensic science, there are practi-cal contingencies which can be the source of difR cul-ties or even interpretation errors. An important con-tingency is the quantity of the evidential material. Any living cell contains about 6 pg of DNA, and the main forensic DNA sources contain about 30 ng
L\1(blood), 400 ngL\1(semen), 2 ngL\1(saliva)
and 0}250 ng per hair root. As indicated in Table 2. RFLP typing would require at least a few microlitres of blood and the various PCR methods require about 0.05L. The theoretical PCR sensitivity of one mol-ecule would suggest that less than 1 nL of blood should be sufRcient. However, this does not take into account the potential loss during DNA isolation, or, more importantly, that one of the alleles may be absent or underepresented in small samples containing only a few molecules, for simple stochastic reasons. As a consequence, it will not appear at the end of the process: a heterozygote will appear as a homozygote. In other words, there may be an allele drop-out.
Another important factor is the quality of the ma-terial. Although DNA is a robust molecule and usable DNA fragments have been recovered from thousand year old mummiRed bodies, it can very quickly be degraded into smaller fragments when exposed to humidity, heat or sunlight. With such degraded sam-ples, DNA typing methods like STR typing, which analyse very short DNA fragments, may still be ap-plied successfully. However, RFLP typing may be no longer possible. Or even worse, a DNA sample pos-sessing a large and a small allele at a VNTR locus may have larger fragments which are too degraded to be detected while the smaller ones will have survived enough to give a detectable band. Here again, the result is an allele drop-out. That is why, in the usual procedure, a small portion of the sample is loaded on
a minielectrophoresis gel followed by nonspeciRc DNA staining. The examination of the intensity of the staining and the average size of the DNA fragments allows both quantitative and qualitative evaluation of the available DNA. The quality of the material can also affect RFLP typing through the inhibition of the restriction enzyme. One of the expected DNA frag-ments may appear at a position corresponding to a lar-ger size than it should, because the enzyme has failed to cut the restriction site closest to the VNTR efRciently. The experienced analyst usually has no problems avoiding interpretation errors arising from the above-mentioned situations or others (extra bands due to unsufRciently stringent hybridization, incomplete stripping, internal restriction site or bacterial con-tamination, missing bands due to blotting problems, etc.). However, without extensive controls, it may be difRcult to prove the tentative explanation. And the limited amount of evidence material usually pre-cludes repeating an analysis.
The potent identiRcation power of DNA typing makes the establishment of databases for the storage of DNA proRles possible and desirable. New proRles can be compared with stored proRles to identify crim-inals, link unsolved cases, etc. This necessitates a high level of standardization among the laboratories parti-cipating in the information exchange networks. They must at least analyse the same polymorphic loci and, for RFLP typing, they must use the same enzymes and similar analytical protocols. Because of the large choice of polymorphic loci and analytical methods, the minimum degree of coherence is not at all war-ranted unless a strong effort is made between the laboratories to reach a consensus. And a consensus is difRcult to reach in a fast moving Reld where new technologies are constantly being developed.
There is no doubt that DNA typing is the ‘safest’ identiRcation technique ever used in forensic biology. But, because of its power and its consequent impact in a courtroom, it is and will remain under intense scrutiny by the forensic and legal communities. Con-sequently, a lot of attention has been paid to quality control and proRciency testing. There is certainly the opportunity for future improvements and devel-opments in thisReld.
See Colour Plate 75.
See also: II/Electrophoresis: Blotting; Deoxyribonucleic Acid, Theory of Techniques for Separation; sional Polyacrylamide Gel Electrophoresis; One-dimen-sional Sodium Dodecyl Sulphate Polyacrylamide Gel Electrophoresis.
Further Reading
Ballantyne J, Sensabaugh G and Witkowski J (1989)DNA Technology and Forensic Science, Banbury report 32. Cold Spring Harbor: Cold Spring Harbor Laboratory Press.
Burke T, Dolf G, Jeffreys AJ and Wolff R (eds) (1991)DNA Fingerprinting:Approaches and Applications. Basel: Bi-rkhauser Verlag.
DNA Fingerprinting (1991) Symposium paper. Elec-trophoresis12: 101}232.
Farley MA and Harrington JJ (1991)Forensic DNA Tech-nology. Chelsea, MI: Lewis Publishers.
Kirby LT (1990) DNA Fingerprinting:An Introduction. New York: Stockholm Press.
Proceedings of the Second International Symposium on Human IdentiTcation (1991) Madison. WI: Promega Corporation.
Rittner C and Schneider PM (eds) (1992) Advances in Forensic Haemogenetics, vol. 4. Berlin, Heidelberg: Springer-Verlag.
Capillary Electrophoresis
M. Chiari and L. Ceriotti, Institute of Biocatalysis and Molecular Recognition, CNR, Milan, Italy
Copyright^ 2000 Academic Press
Introduction
One of the earliest reports on DNA capillary elec-trophoresis (CE) was by Cohen et al. who demon-strated the high resolution of nucleosides and oligo-nucleotides, in the absence of a sieving gel, by simply trapping the analyte into sodium dodecyl sulfate (SDS) micelles. One year later, in 1988, the same author discussed the separation of DNA restriction fragments in a gel-free environment. TheRrst reports on the use of polyacrylamide gel capillary columns to separate DNA with remarkable efRciency and resolu-tion were presented the same year by Guttmanet al. and Cohenet al. By 1989, gel-Rlled CE was already a well-established technique. These preliminary re-ports were then followed by a Surry of articles de-scribing applications of capillary gel electrophoresis (CGE) to the separation of DNA restriction frag-ments. In spite of the high resolving power and efRciency offered by CGE in the analysis of nucleic acids (15}30 million plates per metre and a
single-base resolution for fragments ranging from 15 to more than 500 bases were reported), the difRculties of producing adequate gel-Rlled capillaries hindered their greater application. Heiger et al. proposed, as early as 1990, the use of polyacrylamide cross-linked with a very low or zero concentration ofN,N -methyl-enebisacrylamide. Strangely enough, the revolution brought about by the use of polymer solutions as sieving matrices was only evident in 1991 as a result of the work of Guttman and Cooke and Grossman and Soane. Since then, hundreds of reports have dem-onstrated the advantages of performing DNA separ-ations in a narrow fused silica capillary. Due to its high resolving power and quantitative capability, CE has been successfully applied to different kinds of DNA analysis, including the following: DNA se-quencing, separation of restriction fragments, poly-merase chain reaction (PCR) products and synthetic oligonucleotides.