organic papers
Acta Cryst.(2005). E61, o2685–o2686 doi:10.1107/S1600536805023019 Lalancette and Thompson C
14H16O
o2685
Acta Crystallographica Section E Structure Reports Online
ISSN 1600-5368
(
)-
trans
-1,2,3,4,4a,9a-Hexahydroanthrone
Roger A. Lalancette* and Hugh W. Thompson
Carl A. Olson Memorial Laboratories, Depart-ment of Chemistry, Rutgers University, Newark, NJ 07102, USA
Correspondence e-mail: [email protected]
Key indicators
Single-crystal X-ray study
T= 296 K
Mean(C–C) = 0.004 A˚
Rfactor = 0.064
wRfactor = 0.162
Data-to-parameter ratio = 14.2
For details of how these key indicators were automatically derived from the article, see http://journals.iucr.org/e.
#2005 International Union of Crystallography Printed in Great Britain – all rights reserved
The title compound, C14H16O, is largely flat and rigid, with
near coplanarity between the ketone and the aromatic ring. The packing includes two intermolecular C—H O C close contacts.
Comment
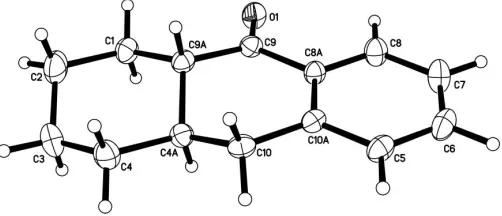
[image:1.610.265.400.382.472.2]The title compound, (I), is the end product in a synthetic sequence designed for a senior chemistry majors’ under-graduate laboratory course (seeExperimental), the final step of which involves acid-catalyzed internal acylation intrans -2-benzylcyclohexanecarboxylic acid (Scribner & Miller, 1965). Both the transand the cisdiastereomers of the product are known, and the equilibrium between them has been shown to favor (I) by a ratio of 88:12 at 298 K (Thompson & Long, 1988). Thus (I) is isolated more easily than thecisepimer, and because (I) is also the higher melting of the pair by some 9 K, it is more easily purified as well.
Fig. 1 shows the asymmetric unit for (I), with its numbering. The molecule has no significant flexibility and is largely flat except for the H atoms; the r.m.s. deviation for all non-H atoms from their average plane is 0.190 (3) A˚ ; the partial flattening of the central ring in turn enforces a slight abnormal flattening in its fully saturated neighbor. Thus, the six internal tetrahedral bond angles for that ring are widened and vary from 110.9 (2) to 112.7 (3), while the ring-carbon torsion
[image:1.610.208.459.610.718.2]Received 14 July 2005 Accepted 19 July 2005 Online 23 July 2005
Figure 1
angles all have absolute values between 51.6 (3) and 56.5 (4),
rather than the 60 expected for cyclohexane. The dihedral
angle for the ketone (C8A/C9/C9A/O1) versus the aromatic ring is 8.69 (11). This coplanarity provides almost the fullest
possible degree of conjugation, reflected in the C O stretching frequency of the IR spectrum (KBr, 1676 cm1).
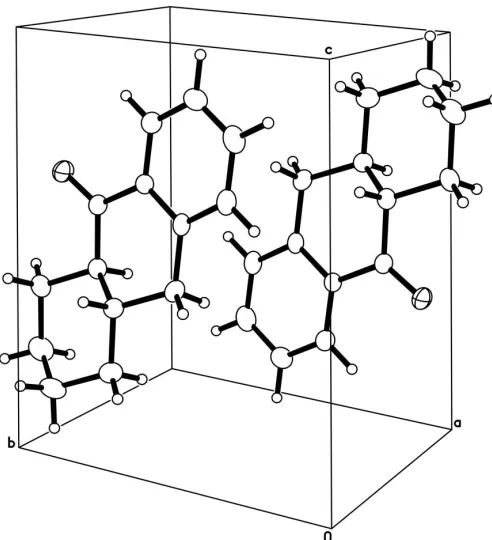
Fig. 2 shows the packing arrangement, involving centro-symmetric pairing of molecules. Despite the short interplanar separations for the aromatic rings (3.87 A˚ within the chosen cell, 3.67 A˚ across thebcface),–stacking is absent because of the offset of the aromatic rings from one another, evident in Fig. 2. Within the 2.7 A˚ range we routinely survey for non-bonded dipolar packing interactions (Steiner, 1997), two intermolecular C—H O C close contacts were found, involving H9A (2.66 A˚ ) in a centrosymmetrically related molecule, and H5A (2.61 A˚ ) in a molecule translationally related alongb(Table 1).
Experimental
Friedel–Crafts acylation of benzene with cis -cyclohexane-1,2-dicar-boxylic acid anhydride (AlCl3) yielded
2-benzoylcyclohexane-carboxylic acid, which was subjected to intentional base-catalyzed stereochemical equilibration to provide thetransepimer (Scribner & Miller, 1965; Lalancette et al., 1996). Subsequent catalytic hydrogenolysis, employing Pd/C in acetic acid, gave trans -2-benzyl-cyclohexanecarboxylic acid, and this was then cyclized at room temperature with H2SO4(Scribner & Miller, 1965). Sublimation of (I)
and recrystallization from hexane provided material of X-ray quality (m.p. 382 K).
Crystal data
C14H16O
Mr= 200.27 Triclinic,P1
a= 7.533 (5) A˚
b= 8.067 (7) A˚
c= 9.317 (5) A˚
= 82.06 (6)
= 80.95 (3)
= 81.31 (7)
V= 548.9 (7) A˚3
Z= 2
Dx= 1.212 Mg m
3
MoKradiation Cell parameters from 25
reflections
= 3.4–9.7
= 0.07 mm1
T= 296 (2) K
Parellelepiped, colourless 0.500.250.07 mm
Data collection
SiemensP4 diffractometer
!/2scans
Absorption correction: numerical (SHELXTL; Sheldrick, 1997)
Tmin= 0.981,Tmax= 0.989 2406 measured reflections 1926 independent reflections 1101 reflections withI> 2(I)
Rint= 0.035
max= 25.0
h=8!1
k=9!9
l=11!11 3 standard reflections
every 97 reflections
intensity decay: variation <2.5%
Refinement
Refinement onF2 R[F2> 2(F2)] = 0.064
wR(F2) = 0.162
S= 1.03 1926 reflections 136 parameters
H-atom parameters constrained
w= 1/[2
(Fo2) + (0.0559P)2 + 0.1357P]
whereP= (Fo2+ 2Fc2)/3 (/)max<0.001
max= 0.13 e A˚
3
min=0.14 e A˚
3
Table 1
Hydrogen-bond geometry (A˚ ,).
D—H A D—H H A D A D—H A
C9A—H9A O1i 0.98 2.66 3.413 (4) 134 C5—H5A O1ii
0.98 2.61 3.354 (4) 133
Symmetry codes: (i)xþ1;yþ2;zþ1; (ii)x;y1;z.
All H atoms were found in electron density difference maps but were placed in calculated positions and allowed to refine as riding on their respective C atoms [C—H = 0.98 A˚ for the aromatic and methine H atoms, C—H = 0.97 A˚ for the methylene H atoms, and
Uiso(H) = 1.2Ueq(C)].
Data collection: XSCANS (Siemens, 1996); cell refinement:
XSCANS; data reduction: XSCANS; program(s) used to solve structure: SHELXTL (Sheldrick, 1997); program(s) used to refine structure:SHELXTL; molecular graphics:SHELXTL; software used to prepare material for publication:SHELXTL.
References
Lalancette, R. A., Cote´, M. L. & Thompson, H. W. (1996).Acta Cryst.C52, 244–246.
Scribner, J. D. & Miller, J. A. (1965).J. Chem. Soc.pp. 5377–5380.
Sheldrick, G. M. (1997).SHELXTL. Version 5.10. Bruker AXS Inc., Madison, Wisconsin, USA.
Siemens (1996).XSCANS. Version 2.2. Siemens Analytical X-ray Instruments Inc., Madison, Wisconsin, USA.
Steiner, T. (1997).Chem. Commun.pp. 727–734.
[image:2.610.315.561.69.339.2]Thompson, H. W. & Long, D. J. (1988).J. Org. Chem.53, 4201–4209.
Figure 2
supporting information
sup-1 Acta Cryst. (2005). E61, o2685–o2686
supporting information
Acta Cryst. (2005). E61, o2685–o2686 [https://doi.org/10.1107/S1600536805023019]
(
±
)-
trans
-1,2,3,4,4a,9a-Hexahydroanthrone
Roger A. Lalancette and Hugh W. Thompson
(±)-trans-1,2,3,4,4a,9a-Hexahydroanthrone
Crystal data
C14H16O Mr = 200.27 Triclinic, P1 Hall symbol: -P 1
a = 7.533 (5) Å
b = 8.067 (7) Å
c = 9.317 (5) Å
α = 82.06 (6)°
β = 80.95 (3)°
γ = 81.31 (7)°
V = 548.9 (7) Å3
Z = 2
F(000) = 216
Dx = 1.212 Mg m−3
Melting point: 382 K
Mo Kα radiation, λ = 0.71073 Å
Cell parameters from 25 reflections
θ = 3.4–9.7°
µ = 0.07 mm−1
T = 296 K
Parellelepiped, colourless 0.50 × 0.25 × 0.07 mm
Data collection
Siemens P4 diffractometer
Radiation source: fine-focus sealed tube Graphite monochromator
2θ/θ scans
Absorption correction: numerical (SHELXTL; Sheldrick, 1997)
Tmin = 0.981, Tmax = 0.989
2406 measured reflections
1926 independent reflections 1101 reflections with I > 2σ(I)
Rint = 0.035
θmax = 25.0°, θmin = 2.2°
h = −8→1
k = −9→9
l = −11→11
3 standard reflections every 97 reflections intensity decay: variation <2.5%
Refinement
Refinement on F2
Least-squares matrix: full
R[F2 > 2σ(F2)] = 0.064 wR(F2) = 0.162
S = 1.03
1926 reflections 136 parameters 0 restraints
Primary atom site location: structure-invariant direct methods
Secondary atom site location: difference Fourier map
Hydrogen site location: inferred from neighbouring sites
H-atom parameters constrained
w = 1/[σ2(F
o2) + (0.0559P)2 + 0.1357P]
where P = (Fo2 + 2Fc2)/3
(Δ/σ)max < 0.001
Δρmax = 0.13 e Å−3
Special details
Experimental. crystal mounted on glass fiber using cyanoacrylate cement
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes.
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2,
conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used
only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2
are statistically about twice as large as those based on F, and R-factors based on ALL data will be even larger.
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
x y z Uiso*/Ueq
O1 0.2615 (3) 0.9955 (2) 0.6283 (2) 0.0638 (7)
C1 0.2729 (4) 1.0875 (3) 0.3302 (3) 0.0608 (9)
H1A 0.3500 1.1529 0.3672 0.073*
H1B 0.1477 1.1303 0.3627 0.073*
C2 0.3074 (6) 1.1091 (4) 0.1631 (4) 0.0851 (12)
H2A 0.2743 1.2266 0.1269 0.102*
H2B 0.4356 1.0787 0.1306 0.102*
C3 0.2000 (5) 1.0009 (4) 0.1016 (3) 0.0840 (12)
H3A 0.2285 1.0132 −0.0042 0.101*
H3B 0.0716 1.0384 0.1264 0.101*
C4A 0.2094 (4) 0.7881 (3) 0.3264 (3) 0.0506 (7)
H4AA 0.0795 0.8183 0.3579 0.061*
C4 0.2413 (5) 0.8169 (4) 0.1608 (3) 0.0677 (9)
H4A 0.1657 0.7513 0.1223 0.081*
H4B 0.3669 0.7768 0.1268 0.081*
C5 0.2265 (4) 0.4159 (3) 0.6212 (4) 0.0582 (8)
H5A 0.2174 0.3234 0.5658 0.070*
C6 0.2182 (5) 0.3862 (4) 0.7704 (4) 0.0711 (10)
H6A 0.2057 0.2725 0.8204 0.085*
C7 0.2272 (5) 0.5156 (4) 0.8506 (4) 0.0745 (10)
H7A 0.2192 0.4946 0.9575 0.089*
C8A 0.2567 (4) 0.7041 (3) 0.6288 (3) 0.0456 (7)
C8 0.2475 (4) 0.6738 (3) 0.7796 (3) 0.0623 (9)
H8A 0.2554 0.7658 0.8359 0.075*
C9 0.2747 (4) 0.8783 (3) 0.5567 (3) 0.0477 (7)
C9A 0.3110 (4) 0.9025 (3) 0.3921 (3) 0.0494 (7)
H9A 0.4408 0.8669 0.3645 0.059*
C10A 0.2475 (4) 0.5746 (3) 0.5476 (3) 0.0454 (7)
C10 0.2602 (4) 0.6054 (3) 0.3841 (3) 0.0524 (8)
H10A 0.1810 0.5375 0.3525 0.063*
supporting information
sup-3 Acta Cryst. (2005). E61, o2685–o2686
Atomic displacement parameters (Å2)
U11 U22 U33 U12 U13 U23
O1 0.0923 (17) 0.0402 (11) 0.0620 (13) −0.0120 (10) −0.0122 (11) −0.0122 (9)
C1 0.072 (2) 0.0440 (16) 0.065 (2) −0.0078 (15) −0.0126 (17) 0.0033 (14)
C2 0.112 (3) 0.067 (2) 0.072 (2) −0.019 (2) −0.012 (2) 0.0170 (18)
C3 0.103 (3) 0.093 (3) 0.051 (2) −0.007 (2) −0.018 (2) 0.0084 (18)
C4A 0.0506 (18) 0.0514 (16) 0.0495 (17) −0.0067 (14) −0.0055 (13) −0.0070 (13)
C4 0.077 (2) 0.072 (2) 0.054 (2) −0.0085 (18) −0.0085 (17) −0.0104 (16)
C5 0.057 (2) 0.0399 (16) 0.076 (2) −0.0063 (14) −0.0046 (16) −0.0061 (14)
C6 0.075 (2) 0.0488 (18) 0.082 (3) −0.0103 (17) −0.0008 (19) 0.0115 (17)
C7 0.096 (3) 0.065 (2) 0.053 (2) −0.0027 (19) −0.0027 (18) 0.0068 (16)
C8A 0.0507 (18) 0.0382 (14) 0.0470 (17) −0.0041 (12) −0.0067 (13) −0.0041 (12)
C8 0.081 (2) 0.0476 (17) 0.056 (2) −0.0028 (16) −0.0078 (16) −0.0061 (14)
C9 0.0524 (18) 0.0392 (14) 0.0537 (18) −0.0070 (13) −0.0103 (14) −0.0085 (13)
C9A 0.0498 (18) 0.0427 (15) 0.0554 (18) −0.0072 (13) −0.0065 (14) −0.0041 (13)
C10A 0.0393 (16) 0.0369 (14) 0.0583 (18) −0.0025 (12) −0.0035 (13) −0.0057 (12)
C10 0.0546 (19) 0.0451 (16) 0.0611 (19) −0.0096 (14) −0.0077 (15) −0.0161 (13)
Geometric parameters (Å, º)
O1—C9 1.214 (3) C5—C6 1.371 (4)
C1—C9A 1.526 (4) C5—C10A 1.385 (4)
C1—C2 1.527 (4) C5—H5A 0.9800
C1—H1A 0.9700 C6—C7 1.381 (5)
C1—H1B 0.9700 C6—H6A 0.9800
C2—C3 1.498 (5) C7—C8 1.373 (4)
C2—H2A 0.9700 C7—H7A 0.9800
C2—H2B 0.9700 C8A—C8 1.385 (4)
C3—C4 1.514 (4) C8A—C10A 1.388 (4)
C3—H3A 0.9700 C8A—C9 1.488 (4)
C3—H3B 0.9700 C8—H8A 0.9800
C4A—C10 1.511 (4) C9—C9A 1.505 (4)
C4A—C4 1.514 (4) C9A—H9A 0.9800
C4A—C9A 1.526 (4) C10A—C10 1.499 (4)
C4A—H4AA 0.9800 C10—H10A 0.9700
C4—H4A 0.9700 C10—H10B 0.9700
C4—H4B 0.9700
C9A—C1—C2 111.3 (3) C10A—C5—H5A 119.5
C9A—C1—H1A 109.4 C5—C6—C7 120.3 (3)
C2—C1—H1A 109.4 C5—C6—H6A 119.8
C9A—C1—H1B 109.4 C7—C6—H6A 119.8
C2—C1—H1B 109.4 C8—C7—C6 119.5 (3)
H1A—C1—H1B 108.0 C8—C7—H7A 120.3
C3—C2—C1 111.1 (3) C6—C7—H7A 120.3
C3—C2—H2A 109.4 C8—C8A—C10A 120.4 (3)
C3—C2—H2B 109.4 C10A—C8A—C9 121.0 (2)
C1—C2—H2B 109.4 C7—C8—C8A 120.4 (3)
H2A—C2—H2B 108.0 C7—C8—H8A 119.8
C2—C3—C4 111.2 (3) C8A—C8—H8A 119.8
C2—C3—H3A 109.4 O1—C9—C8A 121.1 (3)
C4—C3—H3A 109.4 O1—C9—C9A 121.7 (2)
C2—C3—H3B 109.4 C8A—C9—C9A 117.3 (2)
C4—C3—H3B 109.4 C9—C9A—C1 112.1 (2)
H3A—C3—H3B 108.0 C9—C9A—C4A 111.1 (2)
C10—C4A—C4 112.1 (2) C1—C9A—C4A 112.5 (2)
C10—C4A—C9A 110.5 (2) C9—C9A—H9A 106.9
C4—C4A—C9A 110.9 (2) C1—C9A—H9A 106.9
C10—C4A—H4AA 107.7 C4A—C9A—H9A 106.9
C4—C4A—H4AA 107.7 C5—C10A—C8A 118.4 (3)
C9A—C4A—H4AA 107.7 C5—C10A—C10 120.8 (3)
C4A—C4—C3 112.7 (3) C8A—C10A—C10 120.9 (2)
C4A—C4—H4A 109.1 C10A—C10—C4A 113.7 (2)
C3—C4—H4A 109.1 C10A—C10—H10A 108.8
C4A—C4—H4B 109.1 C4A—C10—H10A 108.8
C3—C4—H4B 109.1 C10A—C10—H10B 108.8
H4A—C4—H4B 107.8 C4A—C10—H10B 108.8
C6—C5—C10A 121.0 (3) H10A—C10—H10B 107.7
C6—C5—H5A 119.5
C9A—C1—C2—C3 55.2 (4) C8A—C9—C9A—C4A 36.7 (3)
C1—C2—C3—C4 −56.5 (4) C2—C1—C9A—C9 −179.0 (3)
C10—C4A—C4—C3 −177.1 (3) C2—C1—C9A—C4A −53.0 (3)
C9A—C4A—C4—C3 −53.0 (4) C10—C4A—C9A—C9 −56.9 (3)
C2—C3—C4—C4A 56.1 (4) C4—C4A—C9A—C9 178.1 (2)
C10A—C5—C6—C7 1.2 (5) C10—C4A—C9A—C1 176.6 (2)
C5—C6—C7—C8 −1.0 (5) C4—C4A—C9A—C1 51.6 (3)
C6—C7—C8—C8A 0.7 (5) C6—C5—C10A—C8A −1.1 (4)
C10A—C8A—C8—C7 −0.6 (5) C6—C5—C10A—C10 178.8 (3)
C9—C8A—C8—C7 178.7 (3) C8—C8A—C10A—C5 0.8 (4)
C8—C8A—C9—O1 −8.1 (4) C9—C8A—C10A—C5 −178.5 (3)
C10A—C8A—C9—O1 171.3 (3) C8—C8A—C10A—C10 −179.1 (3)
C8—C8A—C9—C9A 171.5 (3) C9—C8A—C10A—C10 1.6 (4)
C10A—C8A—C9—C9A −9.1 (4) C5—C10A—C10—C4A 157.3 (3)
O1—C9—C9A—C1 −16.9 (4) C8A—C10A—C10—C4A −22.8 (4)
C8A—C9—C9A—C1 163.5 (2) C4—C4A—C10—C10A 174.4 (2)
O1—C9—C9A—C4A −143.7 (3) C9A—C4A—C10—C10A 50.1 (3)
Hydrogen-bond geometry (Å, º)
D—H···A D—H H···A D···A D—H···A
supporting information
sup-5 Acta Cryst. (2005). E61, o2685–o2686
C5—H5A···O1ii 0.98 2.61 3.354 (4) 133
![A triclinic polymorph of (E) 2 (4 isobutylphenyl) N′ [1 (4 nitrophenyl)ethylidene]propanohydrazide](data:image/gif;base64,R0lGODlhAQABAIAAAP///wAAACH5BAEAAAAALAAAAAABAAEAAAICRAEAOw==)