organic papers
Acta Cryst.(2006). E62, o339–o341 doi:10.1107/S1600536805042236 Schlueteret al. C
4H5N3
o339
Acta Crystallographica Section EStructure Reports
Online
ISSN 1600-5368
5-Aminopyrimidine
John A. Schlueter,* Russell J. Funk and Urs Geiser
Materials Science Division, Argonne National Laboratory, 9700 South Cass Avenue, Argonne, IL 60439, USA
Correspondence e-mail: [email protected]
Key indicators
Single-crystal X-ray study
T= 298 K
Mean(C–C) = 0.001 A˚
Rfactor = 0.039
wRfactor = 0.130
Data-to-parameter ratio = 17.3
For details of how these key indicators were automatically derived from the article, see http://journals.iucr.org/e.
#2006 International Union of Crystallography
Printed in Great Britain – all rights reserved
In the title compound, C4H5N3, the nearly planar
5-amino-pyrimidine molecule has twofold rotation symmetry. The crystal structure is stabilized by a hydrogen bond between the amino group and the ring N atoms, thus forming a two-dimensional network parallel to theabplane.
Comment
Recently, there has been considerable interest in the use of pyrimidine (pym) as a bridging ligand for the formation of coordination polymers. Strong magnetic couplings can be mediated by -bonded pym ligands. One-, two- and three-dimensional structural motifs have been studied. For example, Cu(NO3)2(pym)(H2O)2 behaves as a uniform S = 12
anti-ferromagnetic chain (Feyerhermet al., 2000; Yasuiet al., 2001). Structurally, the complex Cu(dca)(NO3)(pym)(H2O) (dca =
dicyanamide) is two-dimensional, but magnetically it behaves as a one-dimensional chain, because the magnetic coupling through the 3-atom pym bridges is significantly stronger than that through the 5-atom dca bridges (Manson et al., 2003). Examples of structurally three-dimensional materials include Cu3(dca)6(pym)20.75H2O (Manson et al., 2003) and
Cu(HCO2)2(pym) (Mansonet al., 2005). The former of these
has a large exchange coupling constant (J/kB=69.4 K), while
the latter exhibits long-range magnetic ordering below TN =
2.8 K. Spontaneous magnetization is also observed in the three-dimensional complexesM(dca)2(pym) (M= Fe or Co),
with ordering temperatures of 3.2 and 1.8 K, respectively (Kusakaet al., 2000).
We are interested in increasing the dimensionality in these systems through the use of pyrimidine derivatives with hydrogen-bonding functionalities. Aminopyrimidines (apym) are one promising family that has this capability. 2-Amino-pyrimidine (2-apym) has been used extensively as a ligand in coordination complexes. For example, a novel molecular tube-like structure containing monodentate 2-apym ligands and the relatively rare1,3,5coordination mode for the dca anions has
been reported forM(dca)2(2-apym) (M= Co or Ni) (Jensenet
al., 2000). Monodentate 2-apym ligands are also found in the complex Cu(dca)2(2-apym)2, which forms one-dimensional
dibridged 1,5-dca chains (van Albadaet al., 2000). When
2-apym acts as a bridging ligand, strong exchange coupling
contants can be obtained, e.g. in [Cu4(2-apym)6(-OCH3)2
-(-F)3(F)2](BF4) (J/kB = 274 K) (van Albadaet al., 2003).
Another example of bridging 2-apym ligands is found in the zigzag chain structure of Cu2(acetate)4(2-apym) (Smithet al.,
1991; Blake et al., 2002). We are aware of no coordination complexes derived from 4-apym or 5-apym. The N atoms of the 2-apym and 4-apym derivatives are more sterically hindered than the 5-apym derivative, thus making 5-apym a promising candidate for a bridging ligand. While the crystal structure of the 4-apym ligand has been published (Van Meervelt & Uytterhoeven, 2003), we report here, for the first time, that of 5-apym,viz.(I).
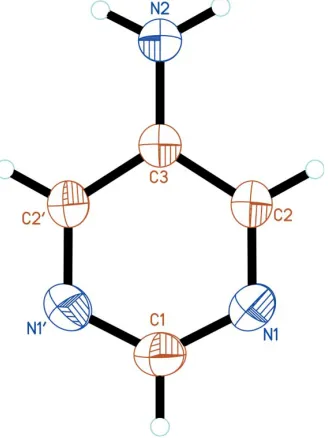
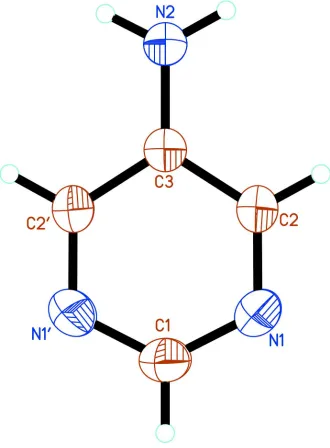
The 5-apym molecule lies on a twofold rotation axis. The atom-numbering scheme is shown in Fig. 1. The bond lengths and angles are typical of pyrimidines, including pym (Wheatley, 1960; Furberget al., 1979), 2-apym (Scheinbeim & Schempp, 1976; Furberget al., 1979) and 4-apym (Van Meer-velt & Uytterhoeven, 2003). The ring atoms deviate only slightly from coplanarity, with atoms N1 and C2 out of the least-squares plane by 0.0022 (6) A˚ . By symmetry, the amino N atom lies in the plane of the pym ring. The dihedral angle between the plane of the amino group and the least-squares plane of the ring is 9.4 (17), signficantly smaller than the 22 angle observed in 2-apym (Scheinbeim & Schempp, 1976).
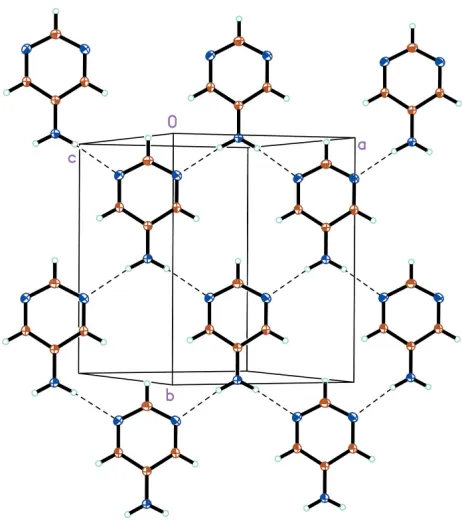
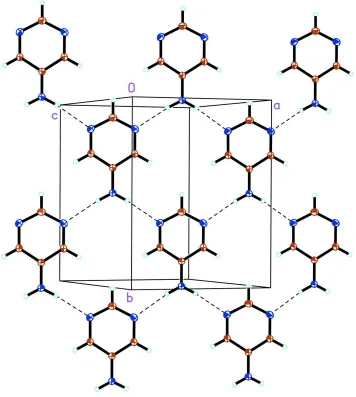
As illustrated in Fig. 2, the packing of the 5-apym molecules is stabilized by an N2—H3 N1i hydrogen bond (Table 2). The hydrogen-bond network results in the formation of two-dimensional sheets parallel to theabplane. The pym plane is tilted by 11.50 (6) with respect to the ab plane. Adjacent sheets are arranged such that uniform slipped stacks of 5-apym molecules form along the a+ bdiagonal. The 5-apym centroid–centroid distance of 3.723 A˚ and perpendicular
separation of 3.362 A˚ confirm the presence of–interactions (Spek, 2003).
Experimental
5-Aminopyrimidine was prepared according to the literature proce-dure of Phillips et al. (1999). Single crystals suitable for X-ray diffraction were grown by recrystallization from benzene.
Crystal data
C4H5N3
Mr= 95.11
Monoclinic,C2=c a= 7.5982 (10) A˚
b= 10.1226 (13) A˚
c= 6.8106 (10) A˚
= 118.596 (5) V= 459.93 (11) A˚3
Z= 4
Dx= 1.374 Mg m
3
MoKradiation Cell parameters from 1724
reflections
= 3.4–29.5
= 0.09 mm1
T= 298 (2) K Rod, colourless 0.500.220.20 mm
Data collection
Siemens SMART CCD area-detector diffractometer
!scans
Absorption correction: integration (XPREPinSHELXTL; Sheldrick, 2001)
Tmin= 0.956,Tmax= 0.987
2534 measured reflections
641 independent reflections 584 reflections withI> 2(I)
Rint= 0.023
max= 29.5
h=10!10
k=14!13
l=9!9
Refinement
Refinement onF2 R[F2> 2(F2)] = 0.039
wR(F2) = 0.130
S= 1.12 641 reflections 37 parameters
H atoms treated by a mixture of independent and constrained refinement
w= 1/[2
(Fo2) + (0.0723P)2
+ 0.0952P]
whereP= (Fo2+ 2Fc2)/3
(/)max= 0.007
max= 0.31 e A˚
3
min=0.17 e A˚
3
Table 1
Selected geometric parameters (A˚ ,).
C1—N1 1.3298 (11)
C2—N1 1.3262 (14)
C2—C3 1.3993 (11)
C3—N2 1.3531 (17)
N1—C1—N1i
125.97 (14)
N1—C2—C3 122.97 (9)
N2—C3—C2 122.68 (6)
C2—C3—C2i
114.65 (12)
C2—N1—C1 116.72 (9)
C3—N2—H3 117.7 (12)
N1—C2—C3—N2 179.77 (6)
N1—C2—C3—C2i
0.23 (6)
C3—C2—N1—C1 0.43 (12) N1i
—C1—N1—C2 0.21 (6)
[image:2.610.94.258.75.294.2]Symmetry code: (i)x;y;zþ1 2.
Table 2
Hydrogen-bond geometry (A˚ ,).
D—H A D—H H A D A D—H A
N2—H3 N1ii 0.89 (2) 2.23 (2) 3.1078 (11) 170 (2)
Symmetry code: (ii)xþ1
2;yþ12;zþ12.
H atoms on aromatic C atoms were positioned geometrically and refined with a riding model, with C—H = 0.93 A˚ . The amino-group H atom was located in a difference map and its position freely refined. For all H atoms,Uiso(H) was constrained to be 1.2 (aromatic) or 1.5 (amino) timesUeqof the carrier atom.
organic papers
o340
Schlueteret al. C4H5N3 Acta Cryst.(2006). E62, o339–o341
Figure 1
[image:2.610.312.558.179.588.2]Data collection:SMART(Bruker, 1997); cell refinement:SAINT
(Bruker, 2001); data reduction: SAINT; program(s) used to solve structure: SHELXTL (Sheldrick, 2001); program(s) used to refine structure:SHELXTL; molecular graphics:SHELXTL; software used to prepare material for publication:SHELXTLandPLATON(Spek, 2003).
Work at Argonne National Laboratory is sponsored by the US Department of Energy, Office of Basic Energy Sciences,
Division of Materials Sciences, under Contract W-31-109-ENG-38. RJF, an undergraduate student at the University of Chicago, is a participant in the US Department of Energy
(DOE) Student Undergraduate Laboratory Research
Internship (SULI) Program, sponsored by the Argonne Division of Educational Programs.
References
Albada, G. A. van, Quiroz-Castro, M. E., Mutikainen, I., Turpeinen, U. & Reedijk, J. (2000).Inorg. Chim. Acta,298, 221–225.
Albada, G. A. van, Roubeau, O., Mutikainen, I., Turpeinen, U. & Reedijk, J. (2003).New J. Chem.1693–1697.
Blake, A. J., Hubberstey, P. & Sampson, C. L. (2002).Acta Cryst.E58, m99– m101.
Bruker (1997).SMART. Version 5.05. Bruker AXS Inc., Madison, Wisconsin, USA.
Bruker (2001).SAINT. Version 6.28a. Bruker AXS Inc., Madison, Wisconsin, USA.
Feyerherm, R., Abens, S., Gu¨nther, D., Ishida, T., Meissner, M., Meschke, M., Nogami, T. & Steiner, M. (2000).J. Phys. Condens. Matter,12, 8495–8509. Furberg, S., Grogaard, J. & Smedsrud, B. (1979).Acta Chem. Scand. B,33, 715–
724.
Jensen, P., Batten, S. R., Moubaraki, B. & Murray, K. S. (2000). Chem. Commun.pp. 793–794.
Kusaka, T., Ishida, T., Hashizume, D., Iwasaki, F. & Nogami T. (2000).Chem. Lett.pp. 1146–1147.
Manson, J. L., Gu, J., Schlueter, J. A. & Wang, H.-H. (2003).Inorg. Chem.42, 3950–3955.
Manson, J. L., Lancaster, T., Chapon, L. C., Blundell, S. J., Schlueter, J. A., Brooks, M. L., Pratt, F. L., Mygren, C. L. & Qualls, J. S. (2005).Inorg. Chem.
44, 989–995.
Phillips, O. A., Murthy, K. S. K., Fiakpui, C. Y. & Knaus, E. E. (1999).Can. J. Chem.77, 216–222.
Scheinbeim, J. & Schempp, E. (1976).Acta Cryst.B32, 607–609.
Sheldrick, G. M. (2001).SHELXTL. Version 6.12. Bruker AXS Inc., Madison, Wisconsin, USA.
Smith, G., Kennard, C. H. L. & Byriel, K. A. (1991).Polyhedron,10, 873–876. Spek, A. L. (2003).J. Appl. Cryst.36, 7–13.
Van Meervelt, L. & Uytterhoeven, K. (2003).Z. Kristallogr. New Cryst. Struct.
218, 481–482.
Wheatley, P. J. (1960).Acta Cryst.13, 80–85.
Yasui, M., Ishikawa, Y., Akiyama, N., Ishida, T., Nogami, T. & Iwasaki, F. (2001).Acta Cryst.B57, 288–295.
organic papers
Acta Cryst.(2006). E62, o339–o341 Schlueteret al. C
[image:3.610.53.284.69.329.2]4H5N3
o341
Figure 2supporting information
sup-1
Acta Cryst. (2006). E62, o339–o341supporting information
Acta Cryst. (2006). E62, o339–o341 [doi:10.1107/S1600536805042236]
5-Aminopyrimidine
John A. Schlueter, Russell J. Funk and Urs Geiser
S1. Comment
Recently, there has been considerable interest in the use of pyrimidine (pym) as a bridging ligand for the formation of
coordination polymers. Strong magnetic couplings can be mediated by µ-bonded pym ligands. One-, two- and
three-dimensional structural motifs have been studied. For example, Cu(NO3)2(pym)(H2O)2 behaves as a uniform S = 1/2
antiferromagnetic chain (Feyerherm et al., 2000; Yasui et al., 2001). Structurally, the complex Cu(dca)(NO3)(pym)(H2O)
(dca = dicyanamide) is two-dimensional, but magnetically it behaves as a one-dimensional chain, because the magnetic
coupling through the 3-atom pym bridges is significantly stronger than that through the 5-atom dca bridges (Manson et
al., 2003). Examples of structurally three-dimensional materials include Cu3(dca)6(pym)2·0.75H2O (Manson et al., 2003)
and Cu(HCO2)2(pym) (Manson et al., 2005). The former of these has a large exchange coupling constant (J/kB = −69.4
K), while the latter exhibits long-range magnetic ordering below TN = 2.8 K. Spontaneous magnetization is also observed
in the three-dimensional complexes M(dca)2(pym) (M = Fe or Co), with ordering temperatures of 3.2 and 1.8 K,
respectively (Kusaka et al., 2000).
We are interested in increasing the dimensionality in these systems through the use of pyrimidine derivatives with
hydrogen-bonding functionalities. Aminopyrimidines (apym) are one promising family that has this capability.
2-Amino-pyrimidine (2-apym) has been used extensively as a ligand in coordination complexes. For example, a novel molecular
tube-like structure containing monodentate 2-apym ligands and the relatively rare µ1,3,5 coordination mode for the dca
anions has been reported for M(dca)2(2-apym) (M = Co or Ni) (Jensen et al., 2000). Monodentate 2-apym ligands are also
found in the complex Cu(dca)2(2-apym)2, which forms one-dimensional dibridged µ1,5-dca chains (van Albada et al.,
2000). When 2-apym acts as a bridging ligand, strong exchange coupling contants can be obtained, e.g. in
[Cu4(2-apym)6(µ-OCH3)2(µ-F)3(F)2](BF4) (J/kB = −274 K) (van Albada et al., 2003). Another example of bridging 2-apym ligands
is found in the zigzag chain structure of Cu2(acetate)4(2-apym) (Smith et al., 1991; Blake et al., 2002). We are aware of
no coordination complexes derived from 4-apym or 5-apym. The N atoms of the 2-apym and 4-apym derivatives are
more sterically hindered than the 5-apym derivative, thus making 5-apym a promising candiate for a bridging ligand.
While the crystal structure of the 4-apym ligand has been published (Van Meervelt & Uytterhoeven, 2003), we report
here, for the first time, that of 5-apym, (I).
The 5-apym molecule lies on a twofold axis. The atom-numbering scheme is shown in Fig. 1. The bond lengths and
angles are typical of pyrimidines, including pym (Wheatley, 1960; Furberg et al., 1979), 2-apym (Scheinbeim &
Schempp, 1976; Furberg et al., 1979) and 4-apym (Van Meervelt & Uytterhoeven, 2003). The ring atoms deviate only
slightly from planarity, with atoms N1 and C2 out of the least-squares plane by 0.0022 (6) Å. By symmetry, the amino N
atom lies in the plane of the pym ring. The dihedral angle between the plane of the amino group and the least-squares
supporting information
sup-2
Acta Cryst. (2006). E62, o339–o341As illustrated in Fig. 2, the packing of the 5-apym molecules is stabilized by an N2—H3···N1i hydrogen bond (Table 2).
The hydrogen-bond network results in the formation of two-dimensional sheets parallel to the ab plane. The pym plane is
tilted by 11.50 (6)° with respect to the ab plane. Adjacent sheets are arranged such that uniform slipped stacks of 5-apym
molecules form along the a + b diagonal. The 5-apym centroid–centroid distance of 3.723 Å and perpendicular separation
of 3.362 Å confirm the presence of π–π interactions (Spek, 2003).
S2. Experimental
5-Aminopyrimidine was prepared according to the literature procedure of Phillips et al. (1999). Single crystals suitable
for X-ray diffraction were grown by recrystallization from benzene.
S3. Refinement
H atoms on aromatic C atoms were positioned geometrically and refined with a riding model, with C—H = 0.93 Å.
[Please check added text] The amino-group H atom was located in a difference map and its position fully refined. For all
supporting information
[image:6.610.140.470.72.520.2]sup-3
Acta Cryst. (2006). E62, o339–o341Figure 1
A view of the molecular structure of 5-aminopyrimidine, showing the atom-numbering scheme. Displacement ellipsoids
supporting information
[image:7.610.128.484.72.469.2]sup-4
Acta Cryst. (2006). E62, o339–o341Figure 2
A packing diagram for 5-aminopyrimidine, illustrating the two-dimensional network in the ab plane. Displacement
ellipsoids are drawn at the 20% probability level and H atoms are shown as small spheres of arbitrary radii. Hydrogen
bonds are depicted as dashed lines.
5-Aminopyrimidine
Crystal data
C4H5N3
Mr = 95.11
Monoclinic, C2/c
Hall symbol: -C 2yc
a = 7.5982 (10) Å
b = 10.1226 (13) Å
c = 6.8106 (10) Å
β = 118.596 (5)°
V = 459.93 (11) Å3
Z = 4
F(000) = 200
Dx = 1.374 Mg m−3
Mo Kα radiation, λ = 0.71073 Å
Cell parameters from 1724 reflections
θ = 3.4–29.5°
µ = 0.09 mm−1
T = 298 K
supporting information
sup-5
Acta Cryst. (2006). E62, o339–o341Data collection
Siemens SMART CCD area-detector diffractometer
Radiation source: fine-focus sealed tube Graphite monochromator
ω scans
Absorption correction: integration
XPREP in SHELXTL (Sheldrick, 2001)
Tmin = 0.956, Tmax = 0.987
2534 measured reflections 641 independent reflections 584 reflections with I > 2σ(I)
Rint = 0.023
θmax = 29.5°, θmin = 3.7°
h = −10→10
k = −14→13
l = −9→9
Refinement
Refinement on F2
Least-squares matrix: full
R[F2 > 2σ(F2)] = 0.039
wR(F2) = 0.130
S = 1.12
641 reflections 37 parameters 0 restraints
Primary atom site location: structure-invariant direct methods
Secondary atom site location: difference Fourier map
Hydrogen site location: inferred from neighbouring sites
H atoms treated by a mixture of independent and constrained refinement
w = 1/[σ2(F
o2) + (0.0723P)2 + 0.0952P] where P = (Fo2 + 2Fc2)/3
(Δ/σ)max = 0.007 Δρmax = 0.31 e Å−3 Δρmin = −0.17 e Å−3
Special details
Experimental. The data collection nominally covered over a hemisphere of reciprocal space, by a combination of four
sets of exposures; each set had a different φ angle for the crystal and each exposure covered 0.3° in ω. The
crystal-to-detector distance was 4.508 cm. Coverage of the unique set was 97.8% complete to at least 29.5° in θ and greater than
99% complete to at least 28.2° in θ. Crystal decay was monitored by repeating the initial 50 frames at the end of data
collection and analyzing the duplicate reflections. Decay was found to be less than 1%, and no decay correction was therefore applied.
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes.
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used
only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2
are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger.
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
x y z Uiso*/Ueq
C1 0.0000 0.09944 (15) 0.2500 0.0506 (4)
H1 0.0000 0.0076 0.2500 0.061*
C2 0.13485 (13) 0.29012 (10) 0.21038 (17) 0.0392 (3)
H2 0.2278 0.3344 0.1824 0.047*
C3 0.0000 0.36475 (12) 0.2500 0.0340 (3)
N1 0.13606 (12) 0.15911 (9) 0.21087 (16) 0.0469 (3)
N2 0.0000 0.49842 (12) 0.2500 0.0491 (4)
supporting information
sup-6
Acta Cryst. (2006). E62, o339–o341Atomic displacement parameters (Å2)
U11 U22 U33 U12 U13 U23
C1 0.0480 (8) 0.0327 (7) 0.0746 (10) 0.000 0.0322 (7) 0.000
C2 0.0355 (5) 0.0380 (5) 0.0500 (6) 0.0009 (3) 0.0253 (4) 0.0007 (3)
C3 0.0336 (6) 0.0328 (6) 0.0379 (6) 0.000 0.0190 (5) 0.000
N1 0.0418 (5) 0.0384 (5) 0.0653 (6) 0.0052 (3) 0.0297 (4) −0.0017 (4)
N2 0.0540 (7) 0.0311 (6) 0.0815 (9) 0.000 0.0480 (7) 0.000
Geometric parameters (Å, º)
C1—N1 1.3298 (11) C2—H2 0.9300
C1—H1 0.9300 C3—N2 1.3531 (17)
C2—N1 1.3262 (14) N2—H3 0.885 (18)
C2—C3 1.3993 (11)
N1—C1—N1i 125.97 (14) C3—C2—H2 118.5
N1—C1—H1 117.0 N2—C3—C2 122.68 (6)
N1i—C1—H1 117.0 C2—C3—C2i 114.65 (12)
N1—C2—C3 122.97 (9) C2—N1—C1 116.72 (9)
N1—C2—H2 118.5 C3—N2—H3 117.7 (12)
N1—C2—C3—N2 179.77 (6) C3—C2—N1—C1 0.43 (12)
N1—C2—C3—C2i −0.23 (6) N1i—C1—N1—C2 −0.21 (6)
Symmetry code: (i) −x, y, −z+1/2.
Hydrogen-bond geometry (Å, º)
D—H···A D—H H···A D···A D—H···A
N2—H3···N1ii 0.885 (18) 2.232 (19) 3.1078 (11) 169.8 (16)