organic papers
o1902
Liet al. C13H9N3O2 doi:10.1107/S1600536805016417 Acta Cryst.(2005). E61, o1902–o1903
Acta Crystallographica Section E Structure Reports Online
ISSN 1600-5368
2-(2-Nitrophenyl)-1
H
-benzimidazole
Xue-Mei Li, Li-Ping Du, Ying Li and Shu-Sheng Zhang*
College of Chemistry and Molecular
Engineering, Qingdao University of Science and Technology, 266042 Qingdao, Shandong, People’s Republic of China
Correspondence e-mail: shushzhang@126.com
Key indicators
Single-crystal X-ray study
T= 293 K
Mean(C–C) = 0.002 A˚
Rfactor = 0.044
wRfactor = 0.113
Data-to-parameter ratio = 13.8
For details of how these key indicators were automatically derived from the article, see http://journals.iucr.org/e.
#2005 International Union of Crystallography Printed in Great Britain – all rights reserved
In the title compound, C13H9N3O2, the dihedral angle between the benzimidazole moiety and the benzene ring is 40.08 (6).
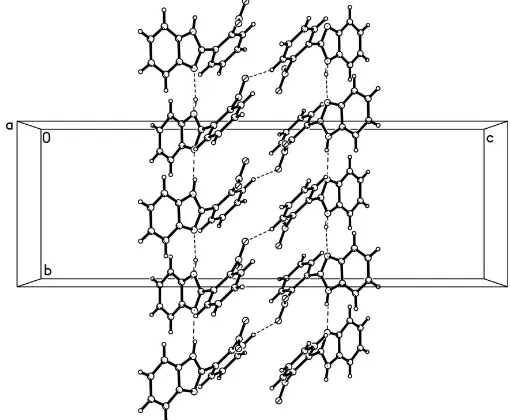
The molecules are linked into chains along the b axis by intermolecular N—H N hydrogen bonds. The chains are interlinked into a two-dimensional network by C—H O hydrogen bonds.
Comment
We have reported the synthesis and crystal structure of 6-methoxycarbonyl-2-methyl-1H-benzimidazol-3-ium nitrate hemihydrate (Ding et al., 2004). In our ongoing studies of benzimidazole derivatives, the title compound, (I), was obtained in the reaction of o-diaminobenzene and 2-nitro-benzoyl chloride.
The bond lengths and angles in (I) are within normal ranges (Allenet al., 1987) and comparable with those in the related compoundN-methyl-2-(o-nitrophenyl)benzimidazole (Das et al., 2003). The benzimidazole moiety is essentially planar, with a dihedral angle of 1.7 (1)between the planes of the benzene
ring and its fused imidazole ring. The whole molecule is non-planar; the benzimidazole ring makes an angle of 40.08 (6)
with the C8–C13 benzene ring. In the crystal structure, the molecules of (I) are linked into chains along thebaxis by N1— H1N N2i intermolecular hydrogen bonds. The chains are interlinked into a two-dimensional network by C12— H12 O1ii hydrogen bonds (Fig. 2; symmetry codes as in Table 2). The packing is further stabilized by van der Waals forces.
Experimental
Compound (I) was synthesized according to the method of Fekneret al.(2004). A solution of 2-nitrobenzoyl chloride (3.71 g, 20 mmol) in CH2Cl2 (40 ml) was added dropwise over 2 h to a solution of
o-diaminobenzene (2.16 g, 20 mmol) and Et3N (3.6 ml) in CH2Cl2
(20 ml). After the addition was complete, the reaction mixture was stirred at 273 K for 1 h and at room temperature for 3 h. The volatiles were removed in vacuo to give an off-white solid. The solid was refluxed in glacial AcOH (50 ml) in the presence of AcONa (1.64 g,
20 mmol) for 15 h. The resulting brown oil was partitioned between CH2Cl2and water. The organic extracts were evaporatedin vacuoto
give a yellow solid. Single crystals of (I) were obtained from a CH2Cl2
solution over a period of 2 d.
Crystal data
C13H9N3O2 Mr= 239.23
Orthorhombic,Pbca a= 7.806 (2) A˚
b= 9.901 (3) A˚
c= 29.307 (8) A˚
V= 2265.1 (11) A˚3 Z= 8
Dx= 1.403 Mg m
3
MoKradiation Cell parameters from 2718
reflections
= 2.8–23.4
= 0.10 mm1 T= 293 (2) K Needle, yellow 0.430.170.12 mm
Data collection
Siemens SMART 1000 CCD area-detector diffractometer
!scans
Absorption correction: multi-scan (SADABS; Sheldrick, 1996)
Tmin= 0.959,Tmax= 0.988
11735 measured reflections
2256 independent reflections 1808 reflections withI> 2(I)
Rint= 0.036
max= 26.2
h=9!9
k=12!10
l=36!32
Refinement
Refinement onF2 R[F2> 2(F2)] = 0.044 wR(F2) = 0.113 S= 1.06 2256 reflections 163 parameters
H-atom parameters constrained
w= 1/[2(F
o2) + (0.0479P)2
+ 0.6337P]
whereP= (Fo2+ 2Fc2)/3
(/)max= 0.001
max= 0.18 e A˚
3
min=0.17 e A˚
3
Table 1
Selected interatomic distances (A˚ ).
N1—C7 1.3509 (19) N1—C1 1.3713 (19)
N2—C6 1.388 (2) N3—C13 1.469 (2)
Table 2
Hydrogen-bond geometry (A˚ ,).
D—H A D—H H A D A D—H A
N1—H1N N2i
0.86 1.99 2.823 (2) 163 C12—H12 O1ii
0.93 2.54 3.285 (3) 137
Symmetry codes: (i)xþ1 2;y
1 2;z; (ii)xþ
1 2;yþ
3 2;z.
After their location in a difference Fourier map, all H atoms were positioned geometrically (N—H = 0.86 A˚ and C—H = 0.93 A˚) and refined as riding, withUiso(H) = 1.2Ueq(parent atom).
Data collection:SMART(Siemens, 1996); cell refinement:SAINT
(Siemens, 1996); data reduction:SAINT; program(s) used to solve structure: SHELXTL (Sheldrick, 1997); program(s) used to refine structure:SHELXTL; molecular graphics:SHELXTL; software used to prepare material for publication:SHELXTL,PARST(Nardelli, 1995) andPLATON(Spek, 2003).
This project was supported by the Programme for New Century Excellent Talents in Universities (grant No. NCET-04-0649) and the Project of Educational Administration of Shandong Province (grant No. J04B12).
References
Allen, F. H., Kennard, O., Watson, D. G., Brammer, L., Orpen, A. G. & Taylor, R. (1987).J. Chem. Soc. Perkin Trans. 2, pp. S1–19.
Das, T. M., Rao, C. P., Kalle, N. & Rissanen, K. (2003).Indian J. Chem. Sect. B,
42, 661–665.
Ding, C. F., Zhang, S. S., Li, X. M., Xu, H. & Ouyang, P. K. (2004).Acta Cryst.
E60, o2441–o2443.
Fekner, T., Gallucci, J. & Chan, M. K. (2004).J. Am. Chem. Soc.126, 223–236. Nardelli, M. (1995).J. Appl. Cryst.28, 659.
Sheldrick, G. M. (1996).SADABS. University of Go¨ttingen, Germany. Sheldrick, G. M. (1997).SHELXTL. Version 5.1. Bruker AXS Inc., Madison,
Wisconsin, USA.
[image:2.610.44.302.68.228.2]Siemens (1996).SMARTandSAINT. Siemens Analytical X-ray Instruments Inc., Madison, Wisconsin, USA.
Figure 1
[image:2.610.311.566.73.283.2]The structure of (I), showing 50% probability displacement ellipsoids and the atom-numbering scheme.
Figure 2
supporting information
sup-1 Acta Cryst. (2005). E61, o1902–o1903
supporting information
Acta Cryst. (2005). E61, o1902–o1903 [https://doi.org/10.1107/S1600536805016417]
2-(2-Nitrophenyl)-1
H
-benzimidazole
Xue-Mei Li, Li-Ping Du, Ying Li and Shu-Sheng Zhang
2-(2-Nitrophenyl)-1H-benzimidazole
Crystal data
C13H9N3O2 Mr = 239.23
Orthorhombic, Pbca Hall symbol: -P 2ac 2ab a = 7.806 (2) Å
b = 9.901 (3) Å c = 29.307 (8) Å V = 2265.1 (11) Å3 Z = 8
F(000) = 992 Dx = 1.403 Mg m−3
Mo Kα radiation, λ = 0.71073 Å Cell parameters from 2718 reflections θ = 2.8–23.4°
µ = 0.10 mm−1 T = 293 K Needle, yellow 0.43 × 0.17 × 0.12 mm
Data collection
Siemens SMART 1000 CCD area-detector diffractometer
Radiation source: fine-focus sealed tube Graphite monochromator
Detector resolution: 8.33 pixels mm-1 ω scans
Absorption correction: multi-scan (SADABS; Sheldrick, 1996) Tmin = 0.959, Tmax = 0.988
11735 measured reflections 2256 independent reflections 1808 reflections with I > 2σ(I) Rint = 0.036
θmax = 26.2°, θmin = 1.4° h = −9→9
k = −12→10 l = −36→32
Refinement
Refinement on F2 Least-squares matrix: full R[F2 > 2σ(F2)] = 0.044 wR(F2) = 0.113 S = 1.06 2256 reflections 163 parameters 0 restraints
Primary atom site location: structure-invariant direct methods
Secondary atom site location: difference Fourier map
Hydrogen site location: inferred from neighbouring sites
H-atom parameters constrained w = 1/[σ2(Fo2) + (0.0479P)2 + 0.6337P]
where P = (Fo2 + 2Fc2)/3 (Δ/σ)max = 0.001
Δρmax = 0.18 e Å−3 Δρmin = −0.17 e Å−3
Special details
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger.
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
x y z Uiso*/Ueq
O1 0.3550 (2) 0.72976 (16) 0.03555 (6) 0.0874 (5) O2 0.17616 (18) 0.88919 (16) 0.04967 (5) 0.0695 (4) N1 0.18779 (16) 0.93846 (12) 0.14460 (4) 0.0385 (3)
H1N 0.2018 0.8540 0.1388 0.046*
N2 0.23100 (17) 1.16064 (12) 0.14295 (5) 0.0399 (3) N3 0.3198 (2) 0.84284 (16) 0.04938 (5) 0.0528 (4) C1 0.06475 (19) 0.99322 (15) 0.17240 (5) 0.0363 (4) C2 −0.0632 (2) 0.93573 (17) 0.19850 (6) 0.0491 (4)
H2 −0.0814 0.8429 0.1988 0.059*
C3 −0.1624 (2) 1.0220 (2) 0.22404 (6) 0.0562 (5)
H3 −0.2497 0.9868 0.2421 0.067*
C4 −0.1351 (2) 1.1611 (2) 0.22348 (6) 0.0542 (5)
H4 −0.2037 1.2163 0.2415 0.065*
C5 −0.0100 (2) 1.21854 (17) 0.19710 (6) 0.0479 (4)
H5 0.0060 1.3116 0.1965 0.057*
C6 0.0925 (2) 1.13270 (14) 0.17111 (5) 0.0370 (4) C7 0.2829 (2) 1.04132 (14) 0.12801 (5) 0.0359 (4) C8 0.4384 (2) 1.02312 (15) 0.10013 (5) 0.0387 (4) C9 0.5800 (2) 1.10263 (19) 0.11062 (7) 0.0557 (5)
H9 0.5704 1.1676 0.1334 0.067*
C10 0.7342 (3) 1.0878 (2) 0.08817 (8) 0.0676 (6)
H10 0.8261 1.1433 0.0956 0.081*
C11 0.7522 (3) 0.9914 (2) 0.05482 (7) 0.0676 (6)
H11 0.8565 0.9808 0.0399 0.081*
C12 0.6160 (3) 0.9109 (2) 0.04355 (7) 0.0573 (5)
H12 0.6275 0.8453 0.0210 0.069*
C13 0.4616 (2) 0.92748 (16) 0.06575 (6) 0.0420 (4)
Atomic displacement parameters (Å2)
U11 U22 U33 U12 U13 U23
supporting information
sup-3 Acta Cryst. (2005). E61, o1902–o1903
C5 0.0567 (11) 0.0336 (9) 0.0534 (11) 0.0094 (8) 0.0028 (9) −0.0050 (7) C6 0.0423 (8) 0.0287 (8) 0.0399 (8) 0.0025 (6) −0.0028 (7) −0.0008 (6) C7 0.0413 (8) 0.0266 (8) 0.0399 (8) −0.0005 (6) −0.0011 (7) −0.0006 (6) C8 0.0411 (8) 0.0305 (8) 0.0445 (9) 0.0014 (7) 0.0001 (7) 0.0014 (7) C9 0.0509 (11) 0.0497 (10) 0.0664 (12) −0.0098 (9) −0.0003 (9) −0.0045 (9) C10 0.0437 (11) 0.0777 (15) 0.0814 (16) −0.0140 (10) −0.0019 (10) 0.0120 (12) C11 0.0432 (10) 0.0870 (16) 0.0725 (14) 0.0102 (11) 0.0146 (10) 0.0124 (12) C12 0.0548 (11) 0.0647 (12) 0.0524 (11) 0.0149 (10) 0.0083 (9) −0.0002 (9) C13 0.0433 (9) 0.0403 (9) 0.0424 (9) 0.0046 (7) 0.0006 (7) 0.0003 (7)
Geometric parameters (Å, º)
O1—N3 1.222 (2) C4—H4 0.93
O2—N3 1.211 (2) C5—C6 1.394 (2)
N1—C7 1.3509 (19) C5—H5 0.93
N1—C1 1.3713 (19) C7—C8 1.474 (2)
N1—H1N 0.86 C8—C9 1.391 (2)
N2—C7 1.3236 (19) C8—C13 1.395 (2)
N2—C6 1.388 (2) C9—C10 1.380 (3)
N3—C13 1.469 (2) C9—H9 0.93
C1—C2 1.381 (2) C10—C11 1.373 (3)
C1—C6 1.398 (2) C10—H10 0.93
C2—C3 1.375 (2) C11—C12 1.370 (3)
C2—H2 0.93 C11—H11 0.93
C3—C4 1.394 (3) C12—C13 1.379 (3)
C3—H3 0.93 C12—H12 0.93
C4—C5 1.369 (3)
C7—N1—C1 107.51 (12) N2—C6—C1 109.50 (13)
C7—N1—H1N 126.2 C5—C6—C1 119.89 (15)
C1—N1—H1N 126.2 N2—C7—N1 112.67 (14)
C7—N2—C6 104.91 (12) N2—C7—C8 123.00 (13)
O2—N3—O1 123.87 (17) N1—C7—C8 124.05 (13)
O2—N3—C13 118.63 (15) C9—C8—C13 116.15 (15)
O1—N3—C13 117.49 (16) C9—C8—C7 117.54 (15)
N1—C1—C2 132.29 (14) C13—C8—C7 126.18 (14)
N1—C1—C6 105.41 (13) C10—C9—C8 121.84 (19)
C2—C1—C6 122.29 (15) C10—C9—H9 119.1
C3—C2—C1 116.94 (16) C8—C9—H9 119.1
C3—C2—H2 121.5 C11—C10—C9 120.17 (19)
C1—C2—H2 121.5 C11—C10—H10 119.9
C2—C3—C4 121.41 (17) C9—C10—H10 119.9
C2—C3—H3 119.3 C12—C11—C10 119.80 (19)
C4—C3—H3 119.3 C12—C11—H11 120.1
C5—C4—C3 121.77 (16) C10—C11—H11 120.1
C5—C4—H4 119.1 C11—C12—C13 119.67 (18)
C3—C4—H4 119.1 C11—C12—H12 120.2
C4—C5—H5 121.2 C12—C13—C8 122.36 (16)
C6—C5—H5 121.2 C12—C13—N3 115.88 (15)
N2—C6—C5 130.58 (14) C8—C13—N3 121.70 (15)
C7—N1—C1—C2 −178.65 (17) N2—C7—C8—C9 37.9 (2) C7—N1—C1—C6 0.15 (17) N1—C7—C8—C9 −135.59 (17) N1—C1—C2—C3 177.78 (17) N2—C7—C8—C13 −146.33 (17) C6—C1—C2—C3 −0.9 (3) N1—C7—C8—C13 40.2 (2) C1—C2—C3—C4 0.1 (3) C13—C8—C9—C10 0.2 (3) C2—C3—C4—C5 0.9 (3) C7—C8—C9—C10 176.38 (17) C3—C4—C5—C6 −1.1 (3) C8—C9—C10—C11 −0.9 (3) C7—N2—C6—C5 178.09 (17) C9—C10—C11—C12 0.7 (3) C7—N2—C6—C1 0.17 (17) C10—C11—C12—C13 0.1 (3) C4—C5—C6—N2 −177.31 (17) C11—C12—C13—C8 −0.8 (3) C4—C5—C6—C1 0.4 (2) C11—C12—C13—N3 176.15 (18) N1—C1—C6—N2 −0.20 (17) C9—C8—C13—C12 0.7 (3) C2—C1—C6—N2 178.75 (15) C7—C8—C13—C12 −175.14 (16) N1—C1—C6—C5 −178.38 (15) C9—C8—C13—N3 −176.15 (16)
C2—C1—C6—C5 0.6 (2) C7—C8—C13—N3 8.0 (3)
C6—N2—C7—N1 −0.08 (17) O2—N3—C13—C12 −145.12 (18) C6—N2—C7—C8 −174.24 (14) O1—N3—C13—C12 33.8 (2) C1—N1—C7—N2 −0.05 (18) O2—N3—C13—C8 31.9 (2) C1—N1—C7—C8 174.05 (14) O1—N3—C13—C8 −149.23 (17)
Hydrogen-bond geometry (Å, º)
D—H···A D—H H···A D···A D—H···A
N1—H1N···N2i 0.86 1.99 2.823 (2) 163
C12—H12···O1ii 0.93 2.54 3.285 (3) 137