doi:10.1251/bpo144 June 20, 2008
Characterizing gene family evolution
David A. Liberles
1#and Katharina Dittmar
1*1Department of Molecular Biology, University of Wyoming, Laramie, WY 82071, USA
#Corresponding Author: David A. Liberles, Department of Molecular Biology, University of Wyoming, Laramie, WY, 82071, USA. Phone: (307) 766 5206;
Fax : (307) 766 5098; Email: [email protected]
*Current Address: Department of Biological Sciences, State University of New York - Buffalo, Buffalo, NY 14260, USA.
Submitted: September 18, 2007; Revised: March 17, 2008; Accepted: April 7, 2008 Indexing terms: genomics; evolution, molecular; phylogeny; sequence homology
ABSTRACT
Gene families are widely used in comparative genomics, molecular evolution, and in systematics. However, they are constructed in different manners, their data analyzed and interpreted differently, with different underlying assumptions, leading to sometimes divergent conclusions. In systematics, concepts like monophyly and the dichotomy between homoplasy and homology have been central to the analysis of phylogenies. We critique the traditional use of such concepts as applied to gene families and give examples of incorrect inferences they may lead to. Operational definitions that have emerged within functional genomics are contrasted with the common formal definitions derived from systematics. Lastly, we question the utility of layers of homology and the meaning of homology at the character state level in the context of sequence evolution. From this, we move forward to present an idealized strategy for characterizing gene family evolution for both systematic and functional purposes, including recent methodological improvements.
INTRODUCTION
As genome scale sequencing has proceeded to generate large datasets of genes from many species, the construction of gene families has become a core activity for both systematics and functional molecular biology. These two pursuits differ not only in their research goals, but also in the terms and concepts used to analyze gene families. The systematics community is concerned with characterizing species relationships through evolution using gene families. The functional molecular biology community is interested in using the evolutionary relationship of genes to understand the details of molecular function. As such, both communities have sets of terms and concepts, with underlying assumptions that are used to characterize the evolutionary process.
METHODS
Gene families as human constructs or as direct
observations of nature
Gene families are a necessary starting point for sequence analysis to understand both functional evolution as well as the systematic relationships of genes and the species they evolved in. Gene families consist of sequences that are collected from various sources, including existing databases and direct sequencing. With these sequences, multiple sequence alignments and phylogenetic trees are generated. The use of terminology becomes controversial here, where the computational and functional communities use words such as construction and generation for the gene families whereas an alternative school of thought might insist that gene families are inherent products of nature and are therefore observed or discovered rather than constructed or generated. This distinction can be important for downstream analysis of
gene families, as an observation that is viewed as deriving from a calculation with underlying assumptions is different from an observation that is presumed to represent the natural order without any assumptions.
Gene families are certainly shaped by natural processes, including speciation, gene duplication, lateral gene transfer, and sequence divergence. However, this evolutionary history is not observed as such, but is rather inferred from sequences. One step in the generation of gene families almost always involves a search for sequence similarity including either a distance threshold or a statement of significance separating an individual gene from one that may have evolved from a different origin to convergently attain sequence similarity. The ultimate aim is to use sequence divergence as an indication of homology, defined here as descent from common ancestry (this is controversial and will be discussed further below). It should be emphasized that an equally important part of the step is to differentiate sequence divergence from sequence convergence.
There are some misunderstandings in this process. As will be discussed below, sequences that are homologous can diverge and subsequently evolve convergently affecting reconstructed tree topologies. Using a broad definition of homology that includes all sequences descended from a common ancestor, this is not a problem. However, sequence similarity can be found among analogous proteins (not descended from a common ancestor) by chance or more probably for functional reasons (1). This becomes a problem only when working on the borders of detectable homology at the sequence level, where protein structures are also involved in the assessment of homology (see for example (2)), and where trees using traditional methods are unlikely to be informative in any regard. Further, this assessment assumes that there were multiple independent origins of genes during the history of life, something that has not been proven (see (3)). The origins of gene families and the assumptions that go into their generation will be important as we move forward.
The term homology is also central to this discussion, which builds upon earlier discussions of the use of the word (4). The origin of the word homology is morphological and refers to common structures. Its utility in modern biology stems from the supposed common origin of such morphological structures and a redefinition involving decent from common ancestry (4). It is this
definition that we will argue has utility in understanding the evolution of genes in families rather than the alternative definitions that have been presented (see (4)). Orthologs are homologs defined phylogenetically through a last common ancestor that diverged through the process of speciation. This definition has nothing to do with function and does not imply that all homologous genes found in different species are orthologs.
Reality and the search for purity
From this starting point, we will move forward with an evaluation of several critical concepts in gene family analysis. Within the systematics community, there has been some intent on a search for conceptual and methodological purity. This includes the view of substitutions occurring site-independently and regularly such that clustering based upon minimizing the number of changes will automatically generate the ancestral tree. However, sequences and the inferences derived from them are dictated by the rules of evolution, which are stochastic, complex, and based upon the behavior of the underlying molecules (proteins and nucleic acids) that govern genomic sequence evolution. Insisting upon conceptual and methodological purity can entail ignoring the process of evolution in its characterization. Concepts such as homology as synapomorphy, layers of homology, homoplasy as implying non-homology, the importance of monophyly, and the importance of 1:1 orthologs in phylogenetic analysis will be discussed in this light. Lastly, we will present a methodological way forward.
Gene family analysis in systematics and the
centrality of monophyly
Monophyly (groups consisting of a common ancestor and all descendants) is a core concept in the systematics community for determining valid taxonomic units ((5); but see also (6)). The importance of monophyly stems from a traditional view of species, where (for the purposes of cataloging the relationships) each monophyletic clade is a true taxonomic entity and is defined by common unique derived character states (referred to as synapomorphies). These are defined by comparison with an outgroup that has an ancestral character state (referred to as the plesiomorphic character state).
level, gene families must be constructed (see above). In evaluating if a gene belongs in a gene family, distance constraints are typically used (for example in single or multiple linkage clustering), with potential modification by the species tree to delimit the oldest node as a speciation event within a particular group of species (7-8). However, The Adaptive Evolution Database (TAED) and other commonly used gene family databases like HOVERGEN (9) have no intrinsic monophyly requirement because of the method of construction. They group sequences that have evolved particularly rapidly along a specific lineage into separate families together with all of the sequences descended from the point of rapid evolution. The reason for this has to do with difficulty in detecting such points using distance methods based upon amino acid divergence. Inclusion of derived amino acid sequences by clustering using synonymous site divergence is conceivable, but is limited in phylogenetic scope.
A fundamental problem with using monophyly at the synonymous site level which may reflect evolutionary history, is that genes that are 100% identical at the amino acid level may in fact not be monophyletic. If one divergent in-group sequence has been subjected to positive diversifying selection based upon amino acid level change in the encoded protein, monophyly at the synonymous site level has no predictive power for assessing gene function. Cases of rapid sequence evolution driving neofunctionalization do occur, especially after gene duplication, but represent a small fraction of total gene family branches (7, 10-11). While difficult to systematically create gene families that reflect evolutionary history, it is desirable to construct such families and then to analyze function in this context. Function is not necessarily monophyletic in that the fate of any given node is probabilistic, dependent upon sequence, fold, and function rather than deterministic (10). This is illustrated below.
The problems with monophyly for gene family
analysis
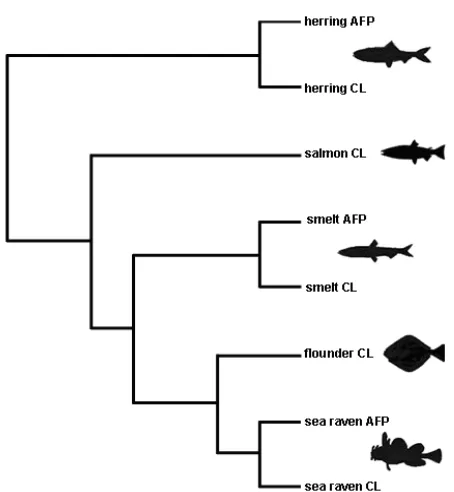
An example of rapid sequence evolution resulting in a non-monophyletic distribution of functions involves the teleost antifreeze proteins (Fig. 1). It is thought that C-type lectins evolved into antifreeze proteins in three independent lineages in teleost evolution (12). Apparently, C-type lectins may have a propensity to
undergo this type of substitution and neofunctionalization compared to other protein folds. Grouping based upon monophyletic clusters of sequences with shared functions would cause the non-monophyletic proteins with shared ancestral sequences and functions (plesiomorphic proteins) to be split into different gene families. In the example in Fig. 1, this would result in all sequences being split away from the family as singletons. For functional genome annotation purposes however, it is clear that conserved plesiomorphic proteins are functionally important to group together in the same family.
Orthologs, paralogs, xenologs and
interpretations of homology
Applied to gene families, the systematic view of homology is almost exclusively used as synonymous with orthology (e.g. orthologous genes), presumably because only these are the genes that have true phylogenetic signal (relating to the history of the organism) through vertical descent (13). This is demonstrated in numerous studies where gene trees are built solely based upon
substitutional information and assumed to be the species tree, without systematic searching or analyzing for duplication or lateral transfer (which is considered to be inappropriate by (13)). Yet, paralogs (the products of gene duplication) and xenologs (the products of lateral transfer) are clearly also descended from a common ancestor (see also (14)) and should not be considered homoplasious noise. Orthologs, paralogs, and xenologs can be examined simultaneously using complex birth/death models that characterize the duplication and lateral transfer processes together with sequence evolution (15-16).
The classification that equates homology exclusively with orthology is problematic in that it does not allow for evolutionary analysis whenever a gene duplication or lateral transfer event has occurred. Given that gene duplication has evidently occurred frequently in many evolutionary lineages (17; reviewed in (18)), it generates problems not only for functional analysis, but also for systematics. Some genes have co-orthologs (2:1 orthologs or in-paralogs) rather than 1:1 orthologs with other species and the lineage-specific gene duplication event would render these co-orthologs as non-homologous according to traditional definitions. The assumption that all sequences are orthologous in systematic analysis is frequently untested to avoid confronting this problem.
Gene family analysis in systematics and
homoplasy
As we move from the gene family level to the character/character state (amino acid or nucleotide) level in gene family analysis we return to the concept of monophyly and its relationship to site evolution. Standard practice in systematics is to infer positional homology of sites (characters) through a multiple sequence alignment and then to build a tree topology from the inferred alignment (19). The tree is used to evaluate patterns of site evolution (character state evolution). Homologous characters are defined by synapomorphic character states (20). These character states emerged from the same evolutionary event and remained identical through evolution. Homology (defined through synapomorphy) is then contrasted with homoplasy, where the same character shows a polyphyletic pattern derived through parallel or convergent evolutionary processes. A character originally identified as homologous (in alignment) that contains a homoplasious character state is then defined as not being homologous (e.g. (21)). While the original
definition of homoplasy was based upon a pattern of independent origin, this has subsequently been extended to mean independent ancestry (see (22) for a discussion).
The problems with interpretations of
homoplasy for gene family analysis
The interpretations above stem from a cladistic view of events. As the molecular evolution and functional genomics communities have increasingly embraced likelihood methods, alternative interpretations of this data and definitions of concepts are used that better characterize the behavior of genes and molecules. Further, while the systematics community has focused on concepts that are useful for classification, the molecular community has focused on concepts that are useful for a mechanistic understanding of evolution (itself potentially important for classification).
The term homology at the character level is operationally used in the molecular community as descent from a common ancestor with modification, consistent with the definition of homology for genes given above. This definition embodies the process of evolution and transitions between character states as a natural probabilistic phenomenon dependent upon the rules of population genetics, molecular biology, and biophysical chemistry.
The advent of likelihood methods has not yet been followed by a linguistic and theoretical framework that embraces evolutionary states with propensities to change (as for example in a Markovian process). The probability to change, whether realized or not, in some evolutionary trajectories, does not change the properties of the state (in a Markovian sense) itself. When an amino acid substitution occurs, it does not make the site where it occurred non-homologous or necessarily functionally different. This view of homology as decent from common ancestry with modification is in direct analogy to the interpretation of evolution at the gene level in constructing gene families.
feature of the evolutionary process, which over a short time frame is governed by the transition probabilities from that state and over a longer time frame by the equilibrium frequencies in the evolutionary process. The concept of layers of homology treats change as a discontinuous process, inconsistent with the expectation of change given the evolutionary model. Further, in this context, the distinction between a homologous position and a nonhomologous character state also fails, by treating evolution as historical independent data points rather than as a scientific process characterized by a statistical model that is supported by a molecular underpinning.
Molecular models are available that characterize the
evolutionary process, grounded in the underlying molecular mechanisms driving evolution (eg (23)). The molecular model might for instance involve differential specificities of different nucleotides for polymerases, the reaction rate of nucleotides with free radicals and under UV light, the enzymatic efficiencies, or specificities of DNA repair enzymes. Farris (24) and Kluge (25) found the concept of differential transition probabilities (such as a transition rate that is different from a transversion rate) to be problematic with regard to this debate as it implied a non-independence of character states and therefore was contentious for a view of homology synonymous with synapomorphy (13, 19-20). Differential transition probabilities create a problem for the character state level of homology (synapomorphy) as some non-identical states are closer to each other than others. However, not only is there strong statistical evidence that the rate of transition is much faster than the rate of transversion, but there is a logical basis for this in nucleic acid biochemistry, in that the transition state of a transversion involves a pyr-pyr or pur-pur intermediate with a high energy distortion to the DNA helix, resulting from a change in the width of the helix itself (see (26) for a review of physical effects on the fidelity of DNA replication). Therefore, it is problematic to use a definition for homology based upon a theoretical framework that is strongly contradicted by well supported models in neighboring fields of science (e.g. biochemistry).
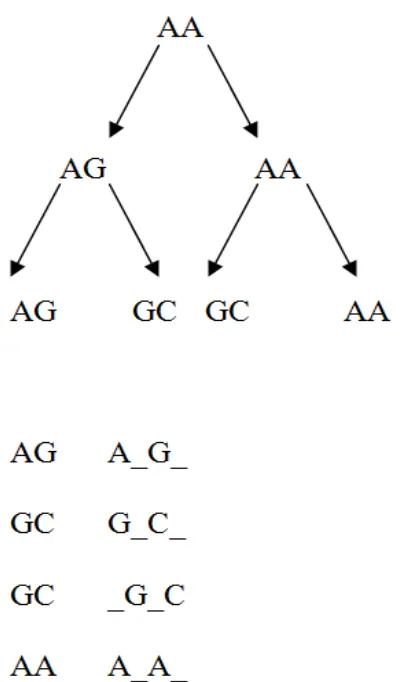
Further, from genomic data, the distinction between homology and homoplasy is artificial, as homoplasy can be observed for homologous characters. Thus, as shown in Fig. 2, a clear case of common ancestry (and thus homology) can be made for the following evolutionary trajectories showing homoplasy. The first nucleotide position underwent parallel evolution and the second involved divergent evolution followed by convergent evolution. At the amino acid level, and especially at the DNA level, numerous characters showing these patterns of evolution can be found involving closely related species. Such patterns are not unexpected for homologous sites, given vertebrate substitution rates and the large number of sites in vertebrate genomes (e.g. TAED (7); HOVERGEN (9)). Further, the molecular procedure based upon the traditional (morphological) homology perception would require homoplasious sites to be placed in different (separate) columns in a multiple sequence alignment, as indicated in the Fig. 3 (20). In fact, this is not common practice for homoplasious sites in molecular
data, as evolutionary models have been used to characterize the process of insertion and deletion, the most common process generating non-homologous positions in an a priori alignment. The evolutionary assessment of insertion and deletion can be done in conjunction with sequence evolution to determine if a homoplasious site is more likely to not be homologous and have arisen by insertion and/or deletion (non-homologous homoplasy) or if the homoplasious site is likely to actually be homologous through shared common ancestry (homologous homoplasy) (27-28). Further, likelihood models are driving this process forward with a definitive statement of homology through simultaneous or iterative alignment and phylogenetic tree calculation to differentiate in a model-based way between homologous homoplasy and non-homologous homoplasy, while considering evolutionary information from gaps (indels)
(29-30). A similar iterative inference can also be made using parsimony (31).
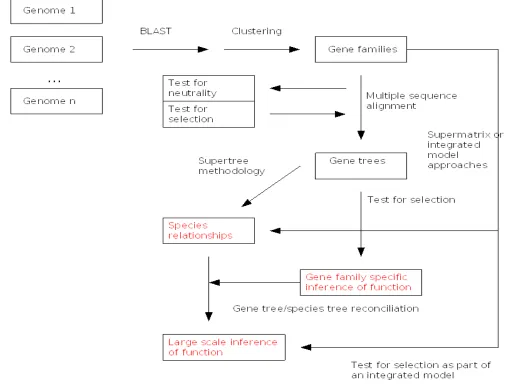
Fig. 3 shows an idealized flow diagram (including controls) for larger scale (genome level) analysis for both systematic and functional purposes as well as smaller scale analysis for functional purposes. Single gene analysis for systematic purposes is sometimes a necessary starting point, but will become more robust with confirmatory evidence from additional genes. While models are necessarily overly simplistic, they are gaining in complexity and realism and are certainly less simplistic and more realistic (albeit less pure) than assuming that Occam’s Razor governs all processes. For example, in addition to integrated models across process levels (discussed above), traditional substitution models (23) have given way to covarion models (32), which are now
moving into models for protein sequence evolution involving structural and functional constraints (33-34). For functional analysis, there have been several recent reviews describing how to use evolutionary information to understand protein function (see for example, (35-36)).
As with any scientific endeavor, controls are necessary, even when using sophisticated models. Tests of neutrality (for models that are based upon neutral stochastic assumptions, like distance, parsimony, and most standard models for maximum likelihood) and tests of saturation (extrapolation of branch lengths over lineages where sites have suffered multiple hits can quickly reach its limits) are critical. This goes beyond not assuming that sequenced genes are orthologs. Scannell et al. (37) have presented a large number of 1:1 paralogs that look like orthologs if not properly tested for. Ultimately, functional and systematic analysis that embraces evolutionary and mechanistic reality rather than philosophical purity will be most accurate.
CONCLUSION
Taken together, strict (non-probabilistic) concepts of monophyly and a characterization of sites showing homoplasy as automatically being non-homologous will be inconsistent with gene family evolution (or evolution in general), and thus also adversely impact our view of species evolution by potentially producing the wrong conclusions. Definitions of homology and homoplasy that are consistent with evolution by descent from common ancestry with modification reflect a useful description of the evolutionary process.
ACKNOWLEDGMENTS
We would like to thank NESCENT for arranging a workshop where these topics were discussed. In particular, we thank Jim Leebens-Mack and Todd Vision as meeting organizers and Mike Sanderson for interesting discussions during the meeting that helped to frame these arguments. Discussions with David Pollock also contributed significantly to this discussion. We thank Mark Gomelsky for comments on the paper as well.
REFERENCES
1. Massey SE, Churbanov A, Rastogi S, Liberles DA. 2008. Characterizing positive and negative selection
and their phylogenetic effects. Gene 2008; 418:22-26. 2. Russell RB, Sasieni PD, Sternberg MJ. Supersites
within superfolds. Binding site similarity in the absence of homology. J Mol Biol 1998; 282:903-918. 3. Britten R. Almost all human genes resulted from
ancient duplication. Proc Natl Acad Sci USA 2006; 103:19027-19032.
4. Fitch WM. Homology: A personal view on some of the problems. Trends in Genetics 2000; 16:227-231.
5. Hennig W. Phylogenetic systematics. Urbana, IL:University of Illinois Press, 1979.
6. Gordon MS. The concept of monophyly: A speculative essay. Biology and Philosophy 1999; 14:331-348.
7. Liberles DA, Schreiber DR, Govindarajan S, Chamberlin SG, Benner SA. The Adaptive Evolution Database (TAED). Genome Biology 2001; 2(8):R0028. 8. Berglund-Sonnhammer AC, Steffansson P, Betts MJ,
Liberles DA. Optimal gene trees from sequences and species trees using a soft interpretation of parsimony.
Journal of Molecular Evolution 2006; 63:240-250.
9. Duret L, Mouchiroud D, Gouy M. HOVERGEN: A database of homologous vertebrate genes. Nucleic
Acids Research 1994; 22:2360-2365.
10. Seoighe C, Johnston CR, Shields DC. Significantly different patterns of amino acid replacement after gene duplication as compared to after speciation. Mol
Biol Evol 2003; 20:484-490.
11. Roth C, Liberles DA. A systematic search for positive selection in higher plants (Embryophytes). BMC Plant
Biology 2006; 6:12.
12. Fletcher GL, Hew CL, Davies PL. Antifreeze proteins of teleost fish. Ann Rev Physiol 2001;63:359-390. 13. Grant T, Kluge AG. Data exploration in phylogenetic
inference: scientific, heuristic, or neither. Cladistics
2003; 19:379-418.
14. Koonin EV. An apology for orthologs- or brave new
memes. Genome Biology 2001;
2(4):comment1005.1-1005.2.
15. Arvestad L, Berglund AC, Lagergren J, Sennblad B. Gene tree reconstruction and orthology analysis based on an integrated model for duplications and sequence evolution. RECOMB 2004; 2004:326-335.
16. Hallett M, Lagergren J, Tofigh A. Simultaneous identification of duplications and lateral transfers.
RECOMB 2004; 2004:347-358.
17. Lynch M, Conery JS. The evolutionary fate and consequences of duplicate genes. Science 2000; 290:1151-1155.
Ekman D, Liberles DA. Evolution after gene duplication: models, mechanisms, sequences, systems, and organisms. J Exp Zool 2007; 308B:58-73.
19. Brower AV, Schawaroch V. Three steps of homology assessment. Cladistics 1996; 12: 265–272.
20. De Pinna MCC. Concepts and test of homology in the cladistic paradigm. Cladistics 1991; 7: 367–394.
21. Page RDM, Holmes EC. Molecular Evolution. A Phylogenetic Approach. Blackwell Publishing, Oxford, 2005.
22. Gould SJ. The structure of evolutionary theory. Cambridge, MA: The Belknap Press of Harvard University Press, 2002.
23. Kimura M. A simple model for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. Journal
of Molecular Evolution 1980; 16:111-120.
24. Farris JS. The logical basis of phylogenetic analysis. In NI Platnick and VA Funk (eds). Advances in Cladistics, vol. 2. New York: Columbia University Press, 1983, pp. 7-36.
25. Kluge AG. Moving targets and shell games. Cladistics
1994; 10:403-413.
26. Kool ET. Hydrogen bonding, base stacking, and steric effects in DNA replication. Ann Rev Biophys Biomol
Struc 2001; 30:1-22.
27. Chang MS, Brenner, SA. Empirical analysis of protein insertions and deletions determining parameters for the correct placement of gaps in protein sequence alignments. J Mol Biol 2004; 341:617-631.
28. Edwards RJ, Shields DC. GASP: Gapped Ancestral Sequence Prediction for proteins. BMC Bioinformatics
2005; 5:123.
29. Lunter G, Miklos I, Drummond A, Jensen JL, Hein J. Bayesian coestimation of phylogeny and sequence alignment. BMC Bioinformatics 2005; 6:83.
30. Redelings BD, Suchard MA. Joint Bayesian estimation of alignment and phylogeny. Syst Biol 2005; 54:401-418.
31. Wheeler WC. Sequence alignment, parameter sensitivity, and the phylogenetic analysis of molecular data. Systematic Biology 1995; 44:321-331.
32. Galtier N. Maximum likelihood phylogenetic analysis under a covarion-like model. Mol Biol Evol 2001; 18:866-873.
33. Depristo MA, Weinreich DM, Hartl DL. Missense meanderings in sequence space: A biophysical view of protein evolution. Nature Reviews Genetics 2005; 6:678-687.
34. Kleinman CL, Rodrigue N, Bonnard C, Philippe H, Lartillot N. A maximum likelihood framework for protein design. BMC Bioinformatics 2006; 7:326.
35. Benner SA. Interpretive proteomics- finding biological meaning in genome and proteome databases. Adv
Enzyme Reg 2003; 43:271-359.
36. Anisimova M, Liberles DA. The quest for natural selection in the age of comparative genomics. Heredity
2007; 99:567-579.
37. Scannell DR, Frank AC, Conant GC, Byrne KP, Woolfit M, Wolfe KH. Independent sorting-out of thousands of duplicated gene pairs in two yeast species descended from a whole genome duplication. Proc
Natl Acad Sci USA 2007; 104:8397-8402.
38. Page RDM. TREEVIEW: An application to display phylogenetic trees on personal computers. Computer