Comparative venom gland transcriptomics
of
Naja kaouthia
(monocled cobra) from
Malaysia and Thailand: elucidating
geographical venom variation and
insights into sequence novelty
Kae Yi Tan1, Choo Hock Tan2, Lawan Chanhome3and Nget Hong Tan1 1Department of Molecular Medicine, Faculty of Medicine, University of Malaya, Kuala Lumpur,
Malaysia
2Department of Pharmacology, Faculty of Medicine, University of Malaya, Kuala Lumpur, Malaysia
3Queen Saovabha Memorial Institute, Bangkok, Thailand
ABSTRACT
Background:The monocled cobra (Naja kaouthia) is a medically important venomous snake in Southeast Asia. Its venom has been shown to vary geographically in relation to venom composition and neurotoxic activity, indicating vast diversity of the toxin genes within the species. To investigate the polygenic trait of the venom and its locale-specific variation, we profiled and compared the venom gland transcriptomes ofN. kaouthia from Malaysia (NK-M) and Thailand (NK-T) applying next-generation sequencing (NGS) technology.
Methods:The transcriptomes were sequenced on the Illumina HiSeq platform, assembled and followed by transcript clustering and annotations for gene expression and function. Pairwise or multiple sequence alignments were conducted on the toxin genes expressed. Substitution rates were studied for the major toxins co-expressed in NK-M and NK-T.
Results and discussion:The toxin transcripts showed high redundancy (41–82% of the total mRNA expression) and comprised 23 gene families expressed in NK-M and NK-T, respectively (22 gene families were co-expressed). Among the venom genes, three-finger toxins (3FTxs) predominated in the expression, with multiple sequences noted. Comparative analysis and selection study revealed that 3FTxs are genetically conserved between the geographical specimens whilst demonstrating distinct differential expression patterns, implying gene up-regulation for selected principal toxins, or alternatively, enhanced transcript degradation or lack of transcription of certain traits. One of the striking features that elucidates the inter-geographical venom variation is the up-regulation ofa-neurotoxins (constitutes ∼80.0% of toxin’s fragments per kilobase of exon model per million mapped reads (FPKM)), particularly the long-chaina-elapitoxin-Nk2a (48.3%) in NK-T but only 1.7% was noted in NK-M. Instead, short neurotoxin isoforms were up-regulated in NK-M (46.4%). Another distinct transcriptional pattern observed is the exclusively and abundantly expressed cytotoxin CTX-3 in NK-T. The findings suggested correlation with the geographical variation in proteome and toxicity of the venom, and support the call for optimising antivenom production and use in the region. Submitted3 October 2016
Accepted3 March 2017 Published5 April 2017
Corresponding authors
Kae Yi Tan, kytan_kae@yahoo.com Choo Hock Tan,
tanchoohock@gmail.com
Academic editor Bruno Lomonte
Additional Information and Declarations can be found on page 30
DOI10.7717/peerj.3142
Copyright 2017 Tan et al. Distributed under
Besides, the current study uncovered full and partial sequences of numerous toxin genes fromN. kaouthiawhich have not been reported hitherto; these include N. kaouthia-specificL-amino acid oxidase (LAAO), snake venom serine protease
(SVSP), cystatin, acetylcholinesterase (AChE), hyaluronidase (HYA), waprin, phospholipase B (PLB), aminopeptidase (AP), neprilysin, etc. Taken together, the findings further enrich the snake toxin database and provide deeper insights into the genetic diversity of cobra venom toxins.
Subjects Genetics, Molecular Biology, Toxicology, Zoology
Keywords Venom gland transcriptomics,Naja kaouthia, Monocled cobra, Three-finger toxins,
Toxin sequence, Snake venom, Geographical variation
INTRODUCTION
Snake venoms consist mainly of pharmacologically active components fine-tuned by evolution against the physiological processes that maintain prey homeostasis (Calvete et al., 2009;Casewell et al., 2013). Positive selection and repeated duplication have been implicated in toxin genes of many lineages, reflecting the adaptive contribution of snake venoms to fitness, and as a source of selective pressure that drives the predator–prey ‘arms race’ coevolution (Margres et al., 2013). However, the origin of toxin genes has been disputable and different mechanisms are available to explain the evolution of snake venom (Casewell et al., 2014;Hargreaves et al., 2014;Reyes-Velasco et al., 2015). A common perspective holds that the duplication of genes generating paralogous give rise to multigene families following the ‘birth and death’ mode of evolution; where some gene copies went through extensive neofunctionalisation at accelerated rates, while the non-functional forms are gradually lost through degradation or transformed into pseudogenes, a process of purifying selection that further preserves the useful toxin arsenal (Casewell et al., 2013;Nei, Gu & Sitnikova, 1997;Sunagar et al., 2013;Sunagar & Moran, 2015). The long evolutionary processes give rise to the extreme complexity of snake venom, an ecologically critical phenotype of venomous snakes which also ramifies into the treatment management of snakebite envenomation.
Southeast Asia were often indiscriminately labelled asNaja naja kaouthia,N. naja sputatrix,N. naja siamensis,N. naja atra, etc. The current systematics based on molecular phylogenetics has resolved the issue and clearly regarded the species of monocled cobra asN. kaouthia(Wu¨ster, 1996). The venom proteome ofN. kaouthia had also been studied to some extents over the years (Kulkeaw et al., 2007;Laustsen et al., 2015; Vejayan, Khoon & Ibrahim, 2014). It is noteworthy that a recent study has provided a global comparison on the proteomic details ofN. kaouthiavenoms from three different Southeast Asian regions (Malaysia, Thailand and Vietnam) qualitatively (profiling protein subtypes) and quantitatively (addressing relative abundances of the proteins) (Tan et al., 2015d). Substantial venom variations were noted across the geographical samples; notably the ThaiN. kaouthiavenom contains a much higher amount of long-chain neurotoxin (LNTX,∼33%) as compared to the Malaysian (∼4%) and the Vietnamese specimens (not detected), respectively. The proteomic variation was further demonstrated at functional levels through studies on venom lethality, neuromuscular depressant activity and immunological neutralisation using antivenom (Tan et al., 2016a, 2016b). The molecular diversity and the genetic variability of the toxins in the venom, however have not been comprehensively investigated.
To gain a deeper insight into the geographical variation of N. kaouthiavenom, we applied a comparative transcriptomic approach of the venom glands to delineate the genetic variability of the cobra. De novo assembled venom gland transcriptomes of N. kaouthia from Malaysia (NK-M) and Thailand (NK-T) were investigated (the transcriptomes are natural features) using next-generation sequencing (NGS) technology at the Illumina platform, imparting a paired-end approach as previously described (Aird et al., 2013;Rokyta et al., 2012;Tan et al., 2015a). It is hoped that by correlating the transcriptomic findings to its proteome and to the biological activities of the venom, the study will propel the understanding of the spectrum and the molecular diversity of the venom genes in this species.
METHODS
Snake venom gland preparation
Total RNA extraction and mRNA purification
The dissected venom gland tissues were submerged and homogenised in a 1 ml glass homogeniser with TRIzol solution (Invitrogen, Carlsbad, CA, USA) under sterile conditions. This was followed by the addition of 20% chloroform, centrifugation and RNA-free DNAase I treatment to separate RNA from cellular debris and residual DNA. The isolated RNA was then pelleted with isopropyl alcohol and washed with 75% ethanol. Polyadenylated mRNA (poly(A)+mRNA) was subsequently purified from 20 mg of total RNA using oligo (dT) magnetic beads as per the (Illumina, San Diego, CA, USA) manufacturer’s instructions. Two rounds of poly(A)+mRNA isolation were performed.
cDNA library construction and sequencing
Enriched poly(A)+mRNA isolated from the total venom gland RNA was used for cDNA construction. Following purification, the mRNA isolated was fragmented in standard buffers containing divalent cations (Zn2+) into short fragments, which acted as templates for cDNA synthesis. Random hexamer-primer (N6) was used to synthesise the first-strand cDNA, followed by second-strand cDNA synthesis with the double-stranded cDNA as input material, using second strand buffers, dNTPs, RNase H and DNA polymerase I. Short fragments were purified with QIAquick PCR extraction kit (Qiagen, Valencia, CA, USA) and resolved withEBbuffer for end repair and the addition of single adenine nucleotide to aid in the subsequent ligation of the Illumina adaptors, which contain a single thymine (T) base overhang at their 3′ends. Following the ligation of sequencing adaptors, these short fragments of cDNA were PCR-amplified and electrophoresed on a 1.5–2% TAE agarose gel. From the electrophoretic agarose gel, suitable fragments (200–700 nt) were selected as templates for subsequent PCR amplification. During the QC steps, an Agilent 2100 Bioanalyzer and ABI StepOnePlus Real-Time PCR System were used in quantification and qualification of the sample library. Sequencing of the PCR-amplified library of each sample was accomplished separately in a single lane on the Illumina HiSeqTM2000 platform (Illumina, San Diego, CA, USA) with 100-base-pair,
paired-end reads.
Raw sequence data and filtering
Sequenced data from Illumina HiSeqTM2000 were transformed by base calling into
sequence data, called the raw data or raw reads and were stored in FASTQ format. Prior to the transcriptome assembly, a stringent-filtering process of raw sequencing reads was carried out. The reads with more than 20% bases having a quality score of Q< 10, sequences containing more than 5% of ambiguous nucleotides or those containing adaptor sequences were removed with an in-house filter programme (Filter_fq, BGI), yielding clean data or clean reads.
De novo transcriptome assembly
Trinity programme and were sequentially applied to process the large volumes of RNA-seq reads. In brief, this was based on the algorithm of de Bruijn graph construction which began by aligningk-mers (k= 25), and reads with a certain length of overlap were joined to form linear contigs. The reads were mapped back onto contigs, and by referring to paired-end reads, contigs from the same transcript as well as the distances between them were determined. The contigs were then partitioned into clusters, each of which carried a complete set of de Bruijn graphs (representing the transcriptional complexity at a given gene or locus). The graphs were independently processed to obtain full-length transcripts for alternatively spliced isoforms and to tease apart transcripts that
corresponded to paralogous genes.
Transcript clustering
The transcript sequences generated through Trinity were called Unigenes. Unigenes from the transcriptome assembly were further processed for sequence splicing and redundancy removing with TGI clustering tools (TGICL) version 2.1 to acquire non-redundant (NR) transcripts at the longest possible length. The transcripts were then subjected to family clustering, which resulted in two classes of transcripts: (a) clusters, with a prefix CL and the cluster ID behind as contig; (b) singletons, whose ID was simply left with a prefix of Unigene. In each cluster, there were several transcripts with sequence similarities among them being >70%; while the singletons ‘Unigenes’ lack overlapping with other fragments at the given stringency. The value 70% was used to categorise the assembled sequences based on similarity; sequences similar to each other (may or may not be homologous as having >90% similarity) were grouped under a cluster comprising various contigs.
In the following step, the transcript Unigenes were aligned by BLASTx to protein databases in the priority order of NCBI NR, with a cut-off E < 10-5. Proteins with the highest ranks in the BLASTx results were referred to determine the coding region sequences of the Unigenes, followed by translation into amino acid sequences (using the standard codon table). Hence, both the nucleotide sequences (5′–3′) and amino sequences of the Unigene-coding regions were acquired. Transcript Unigenes unaligned to any of the protein databases were analysed by software named ESTScan (Iseli, Jongeneel & Bucher, 1999) to determine the nucleotide sequence (5′–3′) direction and amino sequence of the predicted coding region. The length of sequences assembled was a criterion for assembly success. To remove redundancy from each cluster, the longest sequence in each cluster was chosen as the transcript, meanwhile the length of scaffold was extended based on overlapping sequences using Phrap assembler (release 23.0)
(http://www.phrap.org). The distributions of the lengths of contigs, scaffolds and
Unigenes were calculated. The N50 length statistics was set at N50 > 500 for assembly success.
Functional annotation of the transcripts
Annotation of the transcripts provides information about the mRNA expressions (see below) and the putative protein functions. For functional annotation, the generated transcript sequences were aligned by BLASTx to protein databases NR (cut-off E < 10-5).
Expression annotation of the transcripts
To determine the transcript abundances for the identified genes, the FPKM method (Mortazavi et al., 2008) was used, computed using the RNA-seq by the expectation maximisation (RSEM) tool incorporated in the assembly programme, Trinity (Li & Dewey, 2011). The formula is shown below:
FPKM of gene A¼ 10
6C
N L=103;
where FPKM is set to be the expression of gene A,Cto be the number of fragments (i.e. reads) that uniquely aligned to gene A,Nto be the total number of fragments (i.e. reads) that uniquely aligned to all genes and L to be the base number in the coding sequence (CDS) of gene A. The FPKM method is able to eliminate the influence of different gene length and sequencing discrepancy on the calculation of gene expression.
Venom gland transcript classification based on toxinology
Data S1(from BLAST analyses) was further studied to determine which transcripts
(Unigenes) could be identified as ‘toxin,’ ‘non-toxin’ and ‘unidentified’ categories. Keywords were used in search-and-find of the subject description for each toxin match. In view that the final translated toxin products are proteins in nature, the encoded amino acid sequences were subjected to a BLASTp search to ascertain homology with the latest known NCBI NR protein database restricted to the taxon Serpentes (as of 1 June 2016). Minute expression of highly similar/conserved sequences exclusive to Viperidae (vipers and pit vipers) detected by the sensitive assay were excluded from the current study for possibility of trace contamination. Transcripts for cellular proteins and house-keeping genes were categorised into ‘non-toxins’ while those without significant hits/matches were classified as ‘unidentified.’ The relative expression (FPKM) of BLAST-annotated venom gland transcriptomic Unigenes (percentage of the three categories), the relative abundance and the diversity of various toxins in percentage of (i) total protein-encoding transcripts and (ii) total toxin-encoding transcripts were determined.
Redundancy of gene families
In addition, the redundancy of genes was assessed by dividing the transcriptional activity level or transcript reads (FPKM) with the total number of transcripts within a cluster or a group of genes. High redundancy indicates high expression level of a gene group.
Sequence alignments
using Jalview software v2.9 (Waterhouse et al., 2009). Pairwise sequence alignment was performed on the full-length co-expressed toxin-encoding transcripts between the two specimens with Mutalin software (Corpet, 1988). Together with the annotated sequences, sequence comparisons were carried out and compiled inData S3.
Codon alignment and determination of substitution rates
The toxins nucleotide CDSs were retrieved from the nucleotide assembly file and aligned
(Data S4). The non-synonymous (Ka) and synonymous (Ks) substitution rates per site
(Ka/Ks) of the co-expressed transcripts were calculated using the ‘KaKs_Calculator 2.0’
(Wang et al., 2009a,2009b,2010). This programme implements several candidate models of codon substitution in a maximum likelihood framework. We used the approximate method, MYN method (a modified version of the Yang–Nielsen method) to estimateKa
andKsvalue with default parameters. The findings were tabulated in a table available in
the file ofData S4.
Availability of supporting data
Sequence data from the venom gland transcriptome of the two NK-M and NK-T have been deposited in National Centre for Biotechnology Information (NCBI) Sequence Read Achieve (http://www.ncbi.nlm.nih.gov/Traces/sra/sra.cgi) under Bioproject:
PRJNA302200(AC ID:SRP066203) (NK-M:SRR2917658; NK-T:SRR2917657) (http://
www.ncbi.nlm.nih.gov/sra/SRP066203).
RESULTS AND DISCUSSION
Transcriptome assemblyIllumina HiSeq 2000 sequencing was employed to sequence the transcriptome of the venom gland of two NK-M and NK-T and the data statistics were summarised as in
Table 1. In NK-M, Trinity (Grabherr et al., 2011) created 145,538 contigs (N50 = 588),
rate of the overall toxin gene transcription may vary from snake to snake (perhaps affected by the condition of the snake and the time of tissue sampling), however, the relative expression of the different toxin genes is stable and can be studied compatibly between the snakes. Meanwhile, the ‘unidentified’ and ‘non-toxin’ transcripts in both transcriptomes exhibited much lower expression levels in spite of their very large number of genes (Fig. 1). The toxin genes were expressed at a high redundancy in the venom glands of both NK-M (6311.78 FPKM/transcript) and NK-T (22901.34 FPKM/transcript), respectively, compared to the non-toxin transcripts (68.18 FPKM/transcript in NK-M and 95.06 FPKM/transcript in NK-T), supporting the finding of extremely high expression of venom genes within a restricted set of families (Data S1). Overall, approximately 58.8% and 18.0% of the total FPKM in NK-M and NK-T, respectively, are unrelated to envenomation. Most of these are housekeeping genes associated with cellular metabolisms and hence transcribed at lower FPKM levels.
Complexity of the toxin transcripts
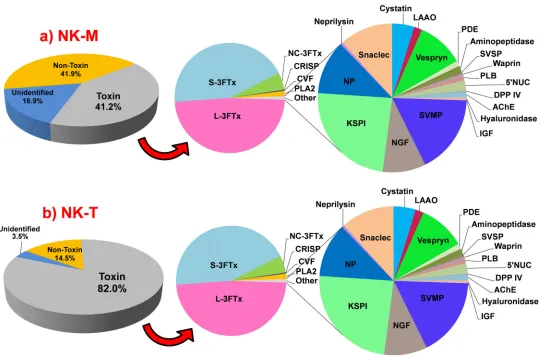
[image:8.612.188.579.120.386.2]The identified toxin transcripts from the venom glands ofN. kaouthiafrom Malaysia (NK-M, 64 partial and complete transcripts, 41.2% of the total FPKM) and Thailand (NK-T, 66 partial and complete transcripts, 82.0% of the total FPKM) comprise 24 gene families, with 23 families identified for each specimen while 22 of them were co-expressed in both (Fig. 1). The fact that the 22 gene families were co-expressed in both NK-M and NK-T venom glands indicates that the toxin gene pool of NK-M and NK-T is
Table 1 Overview of the output statistics.The sequencing and the assembly quality of the venom gland transcriptomes of Malaysian (NK-M) and Thai Naja kaouthia (NK-T).
NK-M NK-T
Total raw reads 56,859,800 59,932,334
Total clean reads 53,663,062 55,186,018
Total clean nucleotides (nt) 4,829,675,580 4,966,741,620
Q20 percentage 98.28 98.31
Npercentage 0.00 0.00
GC percentage 45.84 45.75
Contigs created 145,538 125,435
Total length (nt) 50,843,599 40,514,499
Mean length (nt) 349 323
N50 588 547
Unigenes/transcripts assembled 73,451 69,840
Total length (nt) 51,961,752 39,063,742
Mean length (nt) 707 559
N50 1139 876
Unigenes/transcripts assembled (FPKM > 10) 11,819 4,461
Unidentified 5,725 1,576
Non-toxin 6,030 2,819
largely conserved. Of these, 15 protein gene families were newly detected at the
[image:9.612.36.577.83.439.2]transcriptional level ofN. kaouthiaby the present work; these include Kunitz-type serine protease inhibitor (KSPI), natriuretic peptide (NP), neprilysin, snake venom C-type lectin/lectin-like protein (snaclec), phosphodiesterase (PDE), aminopeptidase (AP), snake venom serine protease (SVSP), waprin, phospholipase B (PLB), 5′nucleotidase (5′NUC), dipeptidylpeptidase-IV (DPP-IV), coagulation factor (CF), acetylcholinesterase (AChE), hyaluronidase (HYA) and insulin-like growth factor (IGF). Some of these toxins were detected in the recent global profiling of N. kaouthiavenom (Laustsen et al., 2015; Tan et al., 2015d), and is in agreement with the previous enzymatic study that has clearly detected PDE, protease, 5′NUC, AChE and HYA activities inN. kaouthia venom (Tan & Tan, 1988).
Figure 1 Abundance of transcripts expressed (FPKM, %) in the venom glands ofNaja kaouthiafrom (a) Malaysia, NK-M; (b) Thailand, NK-T.
Toxins transcripts accounted for 41.2% (NK-M) and 82.0% (NK-T) of the total FPKM, respectively. Three-finger toxin (3FTx) is the most abundant toxin family expressed in the two venom glands (NK-M, 86.8%; NK-T, 97.0% of toxin FPKM). Abbreviations: 3FTx, three-finger toxin; L, long-chain; S, short-long-chain; NC, non-conventional; CRISP, cysteine-rich secretory protein; CVF, cobra venom factor; PLA2, phospholipase A2; SVMP, snake venom metalloproteinase; NGF, nerve growth factor; KSPI, Kunitz-type serine proteinase inhibitor; NP, natriuretic peptide; snaclec, C-type lectin/lectin-like protein; LAAO, L-amino acid oxidase; PDE, phosphodiesterase; AP, aminopeptidase; SVSP, snake venom serine protease;
A total of 63 and 62 NR toxin transcripts were derived from NK-M and NK-T venom glands, respectively (Table 2). Of these, 28 (NK-M) and 27 (NK-T) encoded full-length protein-encoding transcripts (defined here as >90% coverage to the protein-encoding region of the annotated protein sequences) (Tables 3and4). These encompass most of the medically relevant toxins of cobra venoms, including neurotoxins (LNTXs, long neurotoxins; SNTXs, short neurotoxins), cytotoxins (CTXs) and phospholipases A2
(PLA2s) of various isoforms; hence, the species-specific database established herein
represents a valuable depot of bioinformation for further structural and functional studies. It was noted that sequencing of full-length protein-encoding transcripts of several larger venom components was hardly achieved in the current study, probably because Trinity, although a top-performing assembler good at estimating transcript isoforms, has limitations in capturing complete transcript sequences (Honaas et al., 2016). To obtain complete sequences of all genes, we suggest that the current assembly algorithm can be revised with possibly the incorporation of newer programming software in the future. On the other hand, it is also noted that the application of BLASTx search against NR database yielded limited matches to N. kaouthia-specific sequences due to the small database available for this species (Data S2AandS2B;Table 5). As outlined in the methodology, the annotation for most of the transcripts was based on sequence homology to toxins of closely related taxa available in the current repository. This first de novo transcriptomic study forN. kaouthiahas successfully uncovered numerous novel toxin sequences and the data is expected to contribute significantly to enriching the species database (Data S3).
The transcriptomes of both NK-M and NK-T venom glands were extremely biased towards three-finger toxin (3FTxs) expressions (86.8%, NK-M; 97.0%, NK-T, in total FPKM of toxins). Similarly high expression levels of 3FTX genes above 80% to >90% of total toxin expression were reported previously from the venom gland transcriptomes of
Table 2 Overview of the numbers of toxin transcripts, full-length sequences and co-expressed toxin genes from the venom gland transcriptomes of Malaysian (NK-M) and ThaiNaja kaouthia(NK-T).
NK-M NK-T
1. Toxin transcripts 64 66
2. Non-redundant toxin transcriptsa 63 62
3. Protein annotated from non-redundant toxin transcriptsb 49 51 4. Full-length coverage transcript from non-redundant toxins transcriptsc 28 27 5. Non-redundant toxin transcripts co-expressed in both NK-M and NK-T (according to
protein accession ID)d
–Total non-redundant toxins transcriptse 46 43
–Total protein subtypes annotatedf 36 36
Notes:
aNumber of toxins transcripts excluding the redundant transcripts.
bNumber of annotated protein from the non-redundant toxins transcripts (one or more transcripts could be annotated
to same protein ID).
cNon-redundant toxins transcripts with full-length coverage (>90%) to the protein-encoding region of the annotated
protein ID.
dNon-redundant toxins transcripts co-expressed in the venom gland transcriptome of both NK-M and NK-T. eNumber of non-redundant toxins transcripts co-expressed in both NK-M and NK-T.
Table 3 List of full-length transcripts (37 in total) obtained from the venom gland transcriptomes of MalaysianNaja kaouthia(NK-M). Protein
family
Code Protein ID Annotated
accession (UniProt/NCBI)
Species A. acid
(fall in mature chain) Mature chain of accession ID Coverage (mature chain) Coverage (%)
3FTx NKM_FTX01 Alpha-elapitoxin-Nk2a P01391 N. kaouthia 71 71 1–71 100
NKM_FTX05 Cobrotoxin P60770 N. atra 62a 62 22–83 100
NKM_FTX06 Cobrotoxin-c P59276 N. kaouthia 61 61 1–61 100
NKM_FTX07 Short neurotoxin SNTX11 Q2VBP1 O. hannah 56a 57 22–77 98.25
NKM_FTX09 Neurotoxin homolog NL1 Q9DEQ3 N. atra 65a 65 22–86 100
NKM_FTX10 Cytotoxin 5 Q98961 N. atra 60a 60 22–81 100
NKM_FTX11 Cardiotoxin 7 Q91996 N. atra 62a 62 17–83 100
NKM_FTX12 Muscarinic toxin-like protein 2
P82463 N. kaouthia 62 65 1–62 95.38
NKM_FTX13 Muscarinic toxin-like protein 3 homolog
A8N286 O. hannah 65a 65 22–86 100
NKM_FTX14 Weak tryptophan-containing neurotoxin
P82935 N. kaouthia 65 65 22–86 100
NKM_FTX15 Three finger toxin-like Q27J50 L. muta 73a 73 21–93 100
PLA2 NKM_PLA01 Acidic phospholipase A2 1 P00596 N. kaouthia 119 119 28–146 100 vNGF NKM_NGF01 Nerve growth factor beta
chain
A59218 N. kaouthia 116 116 131–246 100
KSPI NKM_KPI01 Protease inhibitor P20229 N. naja 57 57 1–57 100
NKM_KPI02 Kunitz-type protease inhibitor 1
V8N7R6 O. hannah 488a 481 26–506 100b
Snaclec NKM_SCL01 C-type lectin BFL-1 Q90WI8 B. fasciatus 137a 137 22–158 100
NKM_SCL02 C-type lectin BFL-2 Q90WI7 B. fasciatus 137a 137 22–158 100
Cystatin NKM_CYS01 Hypothetical protein L345_15265
V8NBS6 O. hannah 81a 74 1–74 100
NKM_CYS02 Cystatin E3P6P4 N. kaouthia 115 115 27–141 100b
LAAO NKM_LAO01 L-amino-acid oxidase A8QL58 N. atra 495a 430 20–449 100b
Vespryn NKM_VES01 Thaicobrin P82885 N. kaouthia 190 108 1–108 100b
PDE NKM_PDE01 Phosphodiesterase U3FAB3 M. fulvius 836a 836 18–853 100
SVSP NKM_SSP01 Serine protease harobin Q5MCS0 H. curtus 223a 232 34–265 96.12
Waprin NKM_WAP01 Scuwaprin-a B5G6G8 O. scutellatus scutellatus
51a 51 22–72 100
PLB NKM_PLB01 Putative
phospholipase B 81b
F8J2D3 D. coronoides 518a 518 36–553 100
DPP-IV NKM_DPP01 Venom
dipeptidylpeptidase-IV
A6MJH7 P. australis 753a 753 1–753 100
AChE NKM_ACE01 Acetylcholinesterase Q92035 B. fasciatus 551a 578 29–579 95.33
IGF NKM_IGF01 Insulin-like growth factor II
V8NR69 O. hannah 161a 161 25–185 100
Notes:
B,Bungarus;D,Drysdalia;H,Hydrophis;L,Lachesis;M,Micrurus;N,Naja;O,Ophiophagus/Oxyuranus;P,Pseudechis. aNovel protein newly reported for theNaja kaouthiaspecies.
the Chinese cobra (N. atra, 95.8%) and Malaysian king cobra (O. hannah, 84.9%) (Jiang et al., 2011;Tan et al., 2015a). This is consistent with the dominance of 3FTx proteins inN. kaouthiavenom from both localities and these toxins constitute the key
Table 4 List of full-length transcripts (44 in total) obtained from the venom gland transcriptomes of ThaiNaja kaouthia(NK-T). Protein
family
Code Protein ID Annotated
accession (UniProt/NCBI)
Species A. acid (fall
in mature chain) Mature chain of Accession ID Coverage (mature chain) Coverage (%)
3FTx NKT_FTX07 Short neurotoxin SNTX11 Q2VBP1 O. hannah 56a 57 22–77 98.25
NKT_FTX08 Neurotoxin homolog NL1 Q9DEQ3 N. atra 65a 65 22–86 100
NKT_FTX12 Cardiotoxin 7 Q91996 N. atra 62a 62 22–83 100
NKT_FTX13 Muscarinic toxin-like protein 2
P82463 N. kaouthia 62 65 1–62 95.38
NKT_FTX16 Three finger toxin-like precursor
Q27J50 L. muta 73a 73 21–93 100
CRISP NKT_CRP02 Cysteine-rich venom protein kaouthin-2
P84808 N. kaouthia 207 213 26–232 97.18
PLA2 NKT_PLA01 Acidic phospholipase A2 1 P00596 N. kaouthia 119 119 28–146 100 vNGF NKT_NGF01 Nerve growth factor beta
chain precursor
A59218 N. kaouthia 116 116 131–246 100
KSPI NKT_KPI01 Protease inhibitor NACI Q5ZPJ7 N. atra 57a 57 25–81 100
NKT_KPI02 Protease inhibitor B2BS84 A. labialis 229a 232 21–252 98.71
NKT_KPI03 Kunitz-type protease inhibitor 1
V8N7R6 O. hannah 488a 481 26–506 100b
Neprilysin NKT_NEP01 Neprilysin-like XP_007436944 P. bivittatus 672a 672 77–748 100
Snaclec NKT_SCL01 C-type lectin BFL-1 Q90WI8 B. fasciatus 137a 137 22–158 100
NKT_ SCL03 C-type lectin BFL-2 Q90WI7 B. fasciatus 137a 137 22–158 100
Cystatin NKT_CYS01 Hypothetical protein L345_15265
V8NBS6 O. hannah 81a 74 1–74 100b
NKT_CYS02 Cystatin E3P6P4 N. kaouthia 115 115 27–141 100
LAAO NKT_LAO01 L-amino-acid oxidase A8QL58 N. atra 495a 430 20–449 100b
Vespryn NKT_VES01 Thaicobrin P82885 N. kaouthia 190 108 1–108 100b
PDE NKT_PDE01 Phosphodiesterase U3FAB3 M. fulvius 836a 836 18–853 100
SVSP NKT_SSP01 Serine protease harobin Q5MCS0 H. curtus 223a 232 34–265 96.12
Waprin NKT_WAP01 Scuwaprin-a B5G6G8 O. scutellatus
scutellatus
48a 51 25–72 94.12
PLB NKT_PLB01 Putative phospholipase B 81b
F8J2D3 D. coronoides 518a 518 36–553 100
5′NUC NKT_NUC01 Snake venom 5′nucleotidase B6EWW8 G. brevicaudus 524a 524 41–564 100 DPP-IV NKT_DPP01 Venom
dipeptidylpeptidase-IV
A6MJH7 P. australis 753a 753 1–753 100
AChE NKT_ACE01 Acetylcholinesterase Q92035 B. fasciatus 578a 578 29–606 100
HYA NKT_HYA01 Hyaluronidase A3QVN2 E. ocellatus 426a 426 24–449 100
IGF NKT_IGF01 Insulin-like growth factor I isoform X2
XP_007420002 P. bivittatus 168a 155 1–155 100b
Notes:
A,Austrelaps;B,Bungarus;D,Drysdalia;E,Echis;G,Gloydius;H,Hydrophis;L,Lachesis;M,Micrurus;N,Naja;O,Ophiophagus/Oxyuranus;P,Pseudechis/Python. aNovel protein newly reported for theNaja kaouthiaspecies.
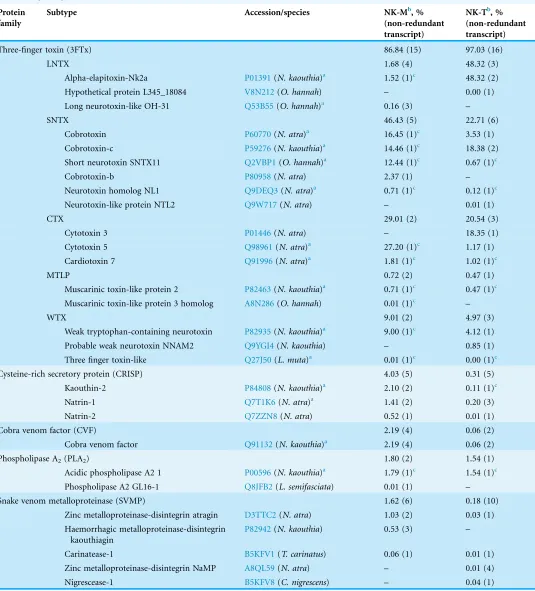
Table 5 Overview of the families and subtypes of toxin genes in the venom gland transcriptomes of Malaysian (NK-M) and Thai Naja kaouthia(NK-T).
Protein family
Subtype Accession/species NK-Mb, %
(non-redundant transcript)
NK-Tb, % (non-redundant transcript)
Three-finger toxin (3FTx) 86.84 (15) 97.03 (16)
LNTX 1.68 (4) 48.32 (3)
Alpha-elapitoxin-Nk2a P01391(N. kaouthia)a 1.52 (1)c 48.32 (2)
Hypothetical protein L345_18084 V8N212(O. hannah) – 0.00 (1)
Long neurotoxin-like OH-31 Q53B55(O. hannah)a 0.16 (3) –
SNTX 46.43 (5) 22.71 (6)
Cobrotoxin P60770(N. atra)a 16.45 (1)c 3.53 (1)
Cobrotoxin-c P59276(N. kaouthia)a 14.46 (1)c 18.38 (2)
Short neurotoxin SNTX11 Q2VBP1(O. hannah)a 12.44 (1)c 0.67 (1)c
Cobrotoxin-b P80958(N. atra) 2.37 (1) –
Neurotoxin homolog NL1 Q9DEQ3(N. atra)a 0.71 (1)c 0.12 (1)c
Neurotoxin-like protein NTL2 Q9W717(N. atra) – 0.01 (1)
CTX 29.01 (2) 20.54 (3)
Cytotoxin 3 P01446(N. atra) – 18.35 (1)
Cytotoxin 5 Q98961(N. atra)a 27.20 (1)c 1.17 (1)
Cardiotoxin 7 Q91996(N. atra)a 1.81 (1)c 1.02 (1)c
MTLP 0.72 (2) 0.47 (1)
Muscarinic toxin-like protein 2 P82463(N. kaouthia)a 0.71 (1)c 0.47 (1)c Muscarinic toxin-like protein 3 homolog A8N286(O. hannah) 0.01 (1)c –
WTX 9.01 (2) 4.97 (3)
Weak tryptophan-containing neurotoxin P82935(N. kaouthia)a 9.00 (1)c 4.12 (1)
Probable weak neurotoxin NNAM2 Q9YGI4(N. kaouthia) – 0.85 (1)
Three finger toxin-like Q27J50(L. muta)a 0.01 (1)c 0.00 (1)c
Cysteine-rich secretory protein (CRISP) 4.03 (5) 0.31 (5)
Kaouthin-2 P84808(N. kaouthia)a 2.10 (2) 0.11 (1)c
Natrin-1 Q7T1K6(N. atra)a 1.41 (2) 0.20 (3)
Natrin-2 Q7ZZN8(N. atra) 0.52 (1) 0.01 (1)
Cobra venom factor (CVF) 2.19 (4) 0.06 (2)
Cobra venom factor Q91132(N. kaouthia)a 2.19 (4) 0.06 (2)
Phospholipase A2(PLA2) 1.80 (2) 1.54 (1)
Acidic phospholipase A2 1 P00596(N. kaouthia)a 1.79 (1)c 1.54 (1)c
Phospholipase A2 GL16-1 Q8JFB2(L. semifasciata) 0.01 (1) –
Snake venom metalloproteinase (SVMP) 1.62 (6) 0.18 (10)
Zinc metalloproteinase-disintegrin atragin D3TTC2(N. atra) 1.03 (2) 0.03 (1)
Haemorrhagic metalloproteinase-disintegrin kaouthiagin
P82942(N. kaouthia) 0.53 (3) –
Carinatease-1 B5KFV1(T. carinatus) 0.06 (1) 0.01 (1)
Zinc metalloproteinase-disintegrin NaMP A8QL59(N. atra) – 0.01 (4)
Nigrescease-1 B5KFV8(C. nigrescens) – 0.04 (1)
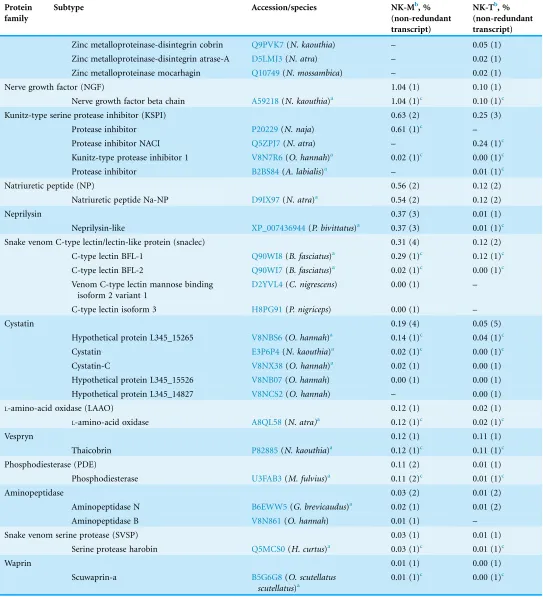
Table 5 (continued ). Protein
family
Subtype Accession/species NK-Mb, %
(non-redundant transcript)
NK-Tb, % (non-redundant transcript)
Zinc metalloproteinase-disintegrin cobrin Q9PVK7(N. kaouthia) – 0.05 (1) Zinc metalloproteinase-disintegrin atrase-A D5LMJ3(N. atra) – 0.02 (1) Zinc metalloproteinase mocarhagin Q10749(N. mossambica) – 0.02 (1)
Nerve growth factor (NGF) 1.04 (1) 0.10 (1)
Nerve growth factor beta chain A59218(N. kaouthia)a 1.04 (1)c 0.10 (1)c
Kunitz-type serine protease inhibitor (KSPI) 0.63 (2) 0.25 (3)
Protease inhibitor P20229(N. naja) 0.61 (1)c –
Protease inhibitor NACI Q5ZPJ7(N. atra) – 0.24 (1)c
Kunitz-type protease inhibitor 1 V8N7R6(O. hannah)a 0.02 (1)c 0.00 (1)c
Protease inhibitor B2BS84(A. labialis)a – 0.01 (1)c
Natriuretic peptide (NP) 0.56 (2) 0.12 (2)
Natriuretic peptide Na-NP D9IX97(N. atra)a 0.54 (2) 0.12 (2)
Neprilysin 0.37 (3) 0.01 (1)
Neprilysin-like XP_007436944(P. bivittatus)a 0.37 (3) 0.01 (1)c
Snake venom C-type lectin/lectin-like protein (snaclec) 0.31 (4) 0.12 (2)
C-type lectin BFL-1 Q90WI8(B. fasciatus)a 0.29 (1)c 0.12 (1)c
C-type lectin BFL-2 Q90WI7(B. fasciatus)a 0.02 (1)c 0.00 (1)c
Venom C-type lectin mannose binding isoform 2 variant 1
D2YVL4(C. nigrescens) 0.00 (1) –
C-type lectin isoform 3 H8PG91(P. nigriceps) 0.00 (1) –
Cystatin 0.19 (4) 0.05 (5)
Hypothetical protein L345_15265 V8NBS6(O. hannah)a 0.14 (1)c 0.04 (1)c
Cystatin E3P6P4(N. kaouthia)a 0.02 (1)c 0.00 (1)c
Cystatin-C V8NX38(O. hannah)a 0.02 (1) 0.00 (1)
Hypothetical protein L345_15526 V8NB07(O. hannah) 0.00 (1) 0.00 (1)
Hypothetical protein L345_14827 V8NCS2(O. hannah) – 0.00 (1)
L-amino-acid oxidase (LAAO) 0.12 (1) 0.02 (1)
L-amino-acid oxidase A8QL58(N. atra)a 0.12 (1)c 0.02 (1)c
Vespryn 0.12 (1) 0.11 (1)
Thaicobrin P82885(N. kaouthia)a 0.12 (1)c 0.11 (1)c
Phosphodiesterase (PDE) 0.11 (2) 0.01 (1)
Phosphodiesterase U3FAB3(M. fulvius)a 0.11 (2)c 0.01 (1)c
Aminopeptidase 0.03 (2) 0.01 (2)
Aminopeptidase N B6EWW5(G. brevicaudus)a 0.02 (1) 0.01 (2)
Aminopeptidase B V8N861(O. hannah) 0.01 (1) –
Snake venom serine protease (SVSP) 0.03 (1) 0.01 (1)
Serine protease harobin Q5MCS0(H. curtus)a 0.03 (1)c 0.01 (1)c
Waprin 0.01 (1) 0.00 (1)
Scuwaprin-a B5G6G8(O. scutellatus
scutellatus)a
venom principles that are important biologically (in predation) and medically (in envenomation) (Tan et al., 2015d). In NK-M, other toxins transcripts include coding cysteine-rich secretory proteins (CRISPs, 4.03%), cobra venom factor (CVF, 2.19%), PLA2s (1.80%), snake-venom metalloproteinases (SVMPs, 1.62%), nerve growth factors
(NGFs, 1.04%) and 17 protein families that were expressed at very low abundance (<1%): KSPIs, NPs, neprilysin, snacles, cystatins,L-amino acid oxidases (LAAOs), vespryn, PDEs,
APs, SVSPs, waprins, PLBs, 5′NUCs, DPP-IV, CFs, AChE and IGFs (Fig. 1). Similarly, the expression of other toxin genes in NK-T venom gland is relatively suppressed: while PLA2s
transcripts charted 1.54% FPKM, and those of the other 21 families are all expressed at a level below 1% FPKM.Table 5shows the comparative gene expression profile of NK-M and NK-T venom glands. The sequences and parametric details of the transcripts were sorted according to gene families and compiled in Data S2AandS2B.
Co-expression of toxin genes between NK-M and NK-T
Table 2shows the key comparison of the venom gland transcriptomes between NK-M
[image:15.612.35.580.102.396.2]and NK-T (also seeData S2A,S2B,S3andTable 5for details). Among the NR toxin transcripts, 46 and 43 transcripts from NK-M and NK-T venom glands, respectively,
Table 5 (continued ). Protein
family
Subtype Accession/species NK-Mb, %
(non-redundant transcript)
NK-Tb, % (non-redundant transcript)
Phospholipase B (PLB) 0.01 (1) 0.01 (1)
Putative phospholipase B 81b F8J2D3(D. coronoides)a 0.01 (1)c 0.01 (1)c
5′nucleotidase (5′NUC) 0.01 (2) 0.02 (1)
5′nucleotidase A6MFL8(D. vestigiata)a 0.00 (1) –
Snake venom 5′nucleotidase F8S0Z7(C. adamanteus) 0.00 (1) – Snake venom 5′nucleotidase B6EWW8(G. brevicaudus)a – 0.02 (1)c
Dipeptidylpeptidase-IV (DPP-IV) 0.01 (1) 0.00 (1)
Venom dipeptidylpeptidase-IV A6MJH7(P. australis)a 0.01 (1)c 0.00 (1)c
Coagulation factor (CF) 0.01 (1) 0.00 (0)
Coagulation factor X isoform 1 V8PHG1(O. hannah) 0.01 (1) –
Acetylcholinesterase (AChE) 0.00 (1) 0.01 (1)
Acetylcholinesterase Q92035(B. fasciatus)a 0.00 (1)c 0.01 (1)c
Hyaluronidase 0.00 (0) 0.01 (2)
Hyaluronidase A3QVN2(E. ocellatus)a – 0.01 (2)c
Insulin-like growth factor (IGF) 0.01 (1) 0.00 (1)
Insulin-like growth factor II V8NR69(O. hannah)a 0.00 (1)c –
Insulin-like growth factor I isoform X2 XP_007420002
(P. bivittatus)
– 0.00 (1)c
Notes:
The number in bracket represents the number of non-redundant transcript.
A,Austrelaps;B,Bungarus;C,Crotalus/Cryptophis;D,Demansia/Drysdalia;E,Echis;G,Gloydius;H,Hydrophis;L,Lachesis/Laticauda;M,Micrurus;N,Naja;
O,Ophiophagus/Oxyuranus;P,Parasuta/Pseudechis/Python;T,Tropidechis.
aToxin transcripts used in the sequence comparative study of NK-M and NK-T (Data S3andS4).
were found to encode the same 36 toxins (Table 2). Of these, selected transcripts with at least 50% coverage of protein-encoding region were compared for sequence similarity (NK-M, 40 transcripts; NK-T, 36 transcripts) (Data S3). In total, 33 pairs of transcripts (including redundancy of allelic variation) encoding 32 different toxin genes were analysed, which encoded 18 of the 24 protein families of toxins (Data S3). The results revealed that most of the transcripts encoding the same toxin are highly conserved between NK-M and NK-T, with only minor divergence at their amino acid sequences
(Data S3). Out of the 33 pairs of sequence comparison (at amino acid level), 29 pairs were
either identical or with <2% divergence; two transcripts varied by 2–10%. Only two transcripts are found to exhibit >10% variation in their amino acid sequences. In theory, the three-dimensional structure of protein is expected to change markedly beyond the ‘twilight zone’ when the amino acid sequence deviates by >30% (Bordoli et al., 2009; Khor et al., 2015). The findings are indicative of a high degree of conserved CDSs in the toxin genes of NK-M and NK-T, and this is supported by results from nucleotide substitution analyses (Data S4), where the nucleotide CDSs of the co-expressed genes were analysed using simple substitution rates analyses program, KaKs_Calculator 2.0. Of note, by analysing the nine pairs of toxins which showed variable sequences, there are no significant findings on positive selection as the mean for genetic differences between the two geographical cobras. The observed sequence conservation in the genes hence may be a result of purifying selection, and the genes are constrained to engage in highly specific interactions with the conserved proteins. Nonetheless, the expression patterns of the conserved genes vary remarkably between NK-M and NK-T, implying complex regulatory processes that the result in the up-regulation of gene-encoding certain toxins, or alternatively, enhanced transcript degradation or lack of transcription that reflects pseudogenisation of certain traits. Variation in the final gene products, i.e. venoms between NK-M and NK-T is indeed conspicuous, where keya-neurotoxins differ remarkably in their relative protein abundance between the two geographical venoms (Tan et al., 2015d). In the present study, the large differences in the principal toxin expression between NK-M and NK-T were most likely the result of directional selection, potentially due to local selective pressures and/or coevolution with distinct prey
populations or species. Adaptive expression variations have been well documented in animals including venomous snakes, where the expression variability has been shown to account for the differences in venom function, particularly intraspecific venom function (Lamichhaney et al., 2015,2016;Margres et al., 2016;Zhang & Reed, 2016).
likely a result of purifying selection. For instance, SVMP and SVSP, typical viperid haemotoxins, are still expressed but at low levels in the venom glands of NK-M and NK-T. This phenomenon of relatively lower expression of SVSP and SVMP in elapids (<5%, in comparison to the levels of 20–30% in the viperid-venom gland transcriptomes) has also been shown in several other elapid-venom gland
transcriptomes (Casewell et al., 2009;Correa-Netto et al., 2011;Jiang et al., 2011; Junqueira-de-Azevedo et al., 2015;Margres et al., 2013;Tan et al., 2015a).
Correlation between transcriptome and proteome
Of the 24 toxin gene families expressed in N. kaouthiavenom gland, only 13 were reported at the protein level as shown in a recent comparative proteomic study (detectable proteins being 3FTx, PLA2, CRISP, SVMP, LAAO, CVF, KSPI, NP, PDE,
5′NUC, vespryn, snaclec and NGF) (Tan et al., 2015d). This shows that a relatively large number of putative toxins were never or only translated and secreted into the venom gland in a minute amount. Several studies have shown that there is no positive correlation between levels of venom-gene expression and protein abundance (Durban et al., 2011;Tan et al., 2015a); although some other studies showed conflicting results (Aird et al., 2013;Casewell et al., 2014;Rokyta, Margres & Calvin, 2015), presumably because of the complex regulation processes governing the mRNA half-life, translation and protein maturation (Vogel & Marcotte, 2012). Admittedly, the issue of correlation between transcriptome and proteome is a complex one (Li, Bickel & Biggin, 2014), as the analysis of the individual gene expression could be further affected by practical factors, e.g. the time span between venom collection and gland tissue harvesting. In this study, the venom glands were sampled four days after venom milking as a way to maximise the yield of total mRNA (Rotenberg, Bamberger & Kochva, 1971), although the different genes might be expressed at different rates.
Three-finger toxins
generally there is lesser isoform variability in these species. Our transcriptomic
results showed that 3FTxs were highly and diversely expressed in bothN. kaouthiavenom glands, with a total of 15 and 16 transcripts identified in NK-M and NK-T, respectively. These transcripts were further classified into long-chain (L-3FTx, represented by LNTX: NK-M, four transcripts; NK-T, three transcripts), short-chain (S-3FTx, including SNTX, CTX and muscarinic toxin-like proteins (MLTPs): NK-M, nine transcripts; NK-T, 10 transcripts) and non-conventional (NC) 3FTxs (NC-3FTx, including weak toxins (WTXs): NK-M, two transcripts; NK-T, three transcripts), according to the classification system based on the number and position of disulphide bonds (Kini & Doley, 2010) (Data S2AandS2B;Fig. 2).
Altogether, a total of 11 and five full-length 3FTx transcripts were uncovered for NK-M and NK-T, respectively (Data S2AandS2B). Among these 3FTx transcripts, seven and four novel sequences were identified for NK-M and NK-T, respectively (Tables 3and4). The CDSs of the 11 3FTx genes that were found co-expressed in both NK-M and NK-T were largely conserved, implying that these paralogous sequences have been maintained by evolution despite potential allopatric subspeciation (Data S4). Although the genetic divergence is lacking within the 3FTxs, the expression levels ofa-neurotoxin genes are noted to vary substantially between NK-M and NK-T (Fig. 1;Table 5). In the NK-M venom gland, S-3FTxs are the dominantly expressed transcripts, whereas in the
NK-T venom gland, L-3FTxs are much more abundantly expressed (Table 5). The findings suggest that the transcription of certain toxin genes may have been selectively suppressed or the up-regulation in other toxin transcripts. A more specific example is the expression of the LNTX,a-elapitoxin-Nk2a (UniProtKB: P01391), a well-investigated long neurotoxin from ThaiN. kaouthiavenom. This unique LNTX gene was found to be co-expressed in both NK-M and NK-T (NKM_FTX01, NKT_FTX01 and NKT_FTX02) but vary significantly between the two at the expression level. Whilea-elapitoxin-Nk2a accounts for 48.3% of toxin gene expression in the NK-T venom gland, its expression is extremely low in the NK-M venom gland, contributing a mere 1.7% of total toxin transcripts. The marked difference in the expression level of this toxin in the respective venom gland of NK-T and NK-M is consistent with its abundance in the venom proteome of NK-T and NK-M (33.3% and 3.9% of total venom protein, respectively) (Tan et al., 2015d). On the other hand, SNTXs are the most abundantly expressed among the transcripts of short-chain 3FTxs (22.68% and 46.39% of toxin gene expression in NK-T and NK-M, respectively), followed by CTXs/cardiotoxins (CTXs, 20.52% and 28.99% in NK-T and NK-M, respectively), whereas MLTPs are expressed at a much lower level in both (MTLPs, 0.47% and 0.71%, respectively in NK-T and NK-M) (Table 5). In contrast to LNTX, SNTXs were expressed at a relatively higher level in NK-M (46.39%) than in NK-T (22.68%). This distinct expression of SNTX was, however, not reflected in the minor difference in SNTX content of the two venoms (4.2% and 7.7% of total venom proteins in NK-M and NK-T, respectively) (Tan et al., 2015d).
(UniProtKB: P59276) from N. kaouthia(Meng et al., 2002;Yang, Yang & Huang, 1969). It is interesting to note that all three cobrotoxins annotated (the former two transcripts, and NKM_FTX08 that is homologous to cobrotoxin-b; UniProtKB: P80958)
Figure 2 Multiple sequence alignment of three-finger toxin (3FTx) transcripts from the venom gland transcriptomes of NK-M and NK-T.
have been previously reported in the venom of N. kaouthia from Yunnan, China (Meng et al., 2002; Qiumin et al., 2002), indicating that the cobrotoxin-encoding gene is well conserved inN. kaouthiafrom distant geographical regions. These SNTXs are highly lethal a-neurotoxins in rodents although some studies suggested that cobra venom SNTXs exhibit a weaker affinity towards the nicotinic acetylcholine receptor (nAChR) as compared to LNTXs, due to the lack of the fifth disulphide bridge in its structure (Barber, Isbister & Hodgson, 2013; Servent et al., 1997, 2000). The weaker affinity of SNTX towards nAChR, however, does not compromise its lethal potency (having compatible LD50 with LNTX), but has important practical concern
as its poor antigenicity is a contributing factor to low neutralisation efficacy of antivenom against SNTX-predominated venoms (Leong et al., 2015; Tan et al., 2015b, 2016a).
Structurally resembling the SNTXs, CTXs are basic proteins with hydrophobic three-finger loops that can interact with the phospholipid bilayer of cell membranes, thereby mediating cytotoxic effect (Feofanov et al., 2005;Konshina et al., 2011;Osipov et al., 2008; Tan & Tan, 2016). CTX genes are highly expressed inN. kaouthia venom glands, with transcripts constituting 28.99% and 20.52% of the total toxin FPKM in NK-M and NK-T, respectively. Overall, transcripts highly homologous to three different CTX genes were detected: CTX-3 (UniProtKB: P01446), CTX-5 (UniProtKB: Q98961) and CTX-7 (UniProtKB: Q91996). Transcript NKT_FTX10, coding CTX-3, was solely expressed in NK-T transcriptome at a significant level (18.35%), representing a potential unique biological marker from the venom gland transcriptome of NK-T. Besides, NKM_FTX10 and NKT_FTX11, expressed respectively by NK-M and NK-T, are homologous to CTX-5 but sequence variation is noted between the two geographical variants. Meanwhile, NKM_FTX11 and NKT_FTX12, co-expressed in both NK-M and NK-M venom glands, exhibit identical sequences to CTX-7 (reported from N. atra), indicating that this CTX gene is relatively well-conserved withinN. kaouthia from Thailand and Malaysia as well as the Taiwanese N. atra(Data S3;Table 5;Fig. 2C).
Phospholipase A2
A total of two and one PLA2transcripts were identified from NK-M and NK-T
venom glands respectively. Among these, NKM_PLA01 and NKT_PLA01 are the predominantly expressed forms of PLA2and they are homologous to Group-IA acidic
PLA2-1 (UniProtKB: P00596) isolated from the venom of N. kaouthiafrom Thailand
(Joubert & Taljaard, 1980) (Fig. 3). From the sequence obtained, NKT_PLA01 was identical to the previously reported sequence (UniProtKB: P00596), while NKM_PLA01 is highly homologous with only one amino acid differing between the two, at the non-functional-critical site (Data S3;Fig. 3). Snake venom PLA2commonly exists in
multiple isoforms and exhibits great diversity in biological properties (Kini, 2003), however, it has not been easy to pinpoint the residues or segments of PLA2that govern
the pharmacological effects (Doley, Zhou & Kini, 2009). In agreement with the proteomic study and purified toxin characterisation (Tan et al., 2015d,2016a), the predominant form of PLA2inN. kaouthiavenom is the acidic, enzymatic PLA2, shown
with conserved Asp-49 residue in the current study. The acidic Asp-49-PLA2, however,
was not lethal in mice even at a dose of >10–20 times of the median lethal dose of whole venom (Tan et al., 2016a). This is consistent with the report of non-toxic property of various acidic-type PLA2s isolated from the venoms of IndianN. kaouthia(Joubert &
Taljaard, 1980) and PakistaniN. naja(Wong, Tan & Tan, 2016), but diverged markedly from the highly lethal, neutral/basic venom PLA2s characterised for other Southeast
Asian cobras including Naja sumatranaandN. sputatrix(Leong et al., 2015;Tan & Arunmozhiarasi, 1989). The acidic PLA2s hence may serve a secondary role of ancillary
function, for instance, potentiating the toxic actions of other venom components, including CTXs/cardiotoxins (Gasanov, Dagda & Rael, 2014), SVMPs (Bustillo et al., 2015) and weak neurotoxin (Mukherjee, 2010) to enhance tissue damages which are crucial for prey digestion but clinically deleterious as it complicates local tissue necrosis. On the other hand, although the mRNA level of the predominant form of PLA2was
relatively low (∼2%) within the venom gland, the PLA2protein content inN. kaouthia
venom is disproportionately higher (12–14% of total venom proteins) (Laustsen et al., 2015;Tan et al., 2015d). A possible explanation for this is that the mRNA of the PLA2
[image:21.612.46.578.87.179.2]may have longer half-life, and thus a low mRNA level is sufficient to produce the necessary PLA2 protein (Vogel & Marcotte, 2012).
Cysteine-rich secretory protein
Although the role of CRISP in the pathogenesis of snake envenomation remains unclear, its occurrence in a wide range of snake venoms indicates that it may play a certain role in the predator envenomation strategies (Sunagar et al., 2015). In view of the protein’s limited diversity and little evidence of gene duplication, it has been suggested that the CRISP gene family is functionally conserved across most lineages (Vonk et al., 2013). The current study showed that five CRISP transcripts obtained from each of the NK-M and NK-T venom gland were all highly homologous to sequences of known snake venom CRISPs (Fig. 4). Of these, the major CRISP transcript in NK-M venom gland was annotated to kaouthin-2 (UniProtKB: P84808), a protein that has been isolated fromN. kaouthiavenom (unspecified geographical origin) (Osipov et al., 2005), whereas natrin-1 (UniProtKB: Q7T1K6) (Jin et al., 2003), a CRISP isolated fromN. atra
venom (Kunming, China) was highly expressed in the NK-T venom gland (Data S2A
andS2B;Table 5). Even though there are substantial variations in the amino acid
sequences between kaouthin and natrin, the two CRISPs were reported to exhibit similar pharmacological activities such as antagonizing the calcium-activated (KCa) channel, voltage-gated potassium channel (Kv) and calcium release channel/ryanodine receptor (RyR) (Chang et al., 2005;Wang et al., 2006). Although the CRISPs were expressed in a range of 2–4% of total venom proteins inN. kaouthia venom samples (Laustsen et al., 2015;Tan et al., 2015d), the mRNA levels differ by 10-fold between NK-M (4.03%) and NK-T (0.32%) (Table 5). The protein expression of CRISPs also appears to
undergo complex regulation, as multiple proteomic studies uncovered only the presence of natrin-type CRISP in the venoms ofN. kaouthiafrom Malaysia and/or Thailand (Kulkeaw et al., 2007;Laustsen et al., 2015;Tan et al., 2015d). From the literature, the previous reported kaouthin-type CRISP was isolated and sequenced fromN. kaouthiaof an unknown geographical source (Osipov et al., 2005).
Cobra venom factor
[image:22.612.36.578.86.166.2]Cobra venom factor as a non-lethal protein resembles the complement C3 proteins structurally and functionally. Nevertheless, its pathogenic role in snake envenomation has been attributed to increasing vascular permeability and blood flow, thus facilitating venom toxins distribution (Vogel & Fritzinger, 2010). In the present study, four and two partial CVF transcripts were detected from NK-M and NK-T venom glands, respectively.
Although the CVF sequences are incomplete, the assembly of these partial transcripts provides full coverage to the annotated protein (UniProtKB: Q91132) when aligned. Sequence alignment shows that the CVF transcripts from NK-T are identically matched to the CVF isolated from N. naja siamensis(UniProtKB: Q91132), however the
N. siamensiscould be a misnomer ofN. kaouthiafrom Thailand during the time of data depositing (Data S3). While the CVF transcripts from NK-T are identical to the N. siamensisCVF, the sequence of CVF transcripts from NK-M on the other hand differs by 2–10% (Data S3), representing sequence variation probably associated with
geographical differences of the species. Although variations were noted, it is likely that the novel CVF from NK-M is functionally similar to the annotated CVF-Q91132, as the variations detected are rather minor and not selection-driven (Data S4).
Snake venom metalloproteinase
Snake venom metalloproteinases are proteases usually found in abundance in viper and pit viper venoms (Tan et al., 2015c;Tang et al., 2016). This multi-locus gene protein family encodes various protease subtypes that exhibit different pharmacological activities, most of which are associated with haemorrhage and coagulopathy (Fox & Serrano, 2008,2009;Markland & Swenson, 2013). In the present study, multiple SVMP transcripts were assembled from the venom glands of N. kaouthia, and these transcripts encode three and seven different subtypes of SVMPs for NK-M and NK-T, respectively (Table 5). The longer transcripts from NK-M (NKM_SMP01 and NKM_SMP02) and NK-T (NKT_SMP01) were partially matched to zinc metalloproteinase-disintegrin atragin (UniProtKB: D3TTC2) of N. atra(Data S3). The annotated SVMPs genes in this study encode class P-III SVMPs, consistent with the finding as demonstrated at the proteomic level by multiple studies (Kulkeaw et al., 2007;Laustsen et al., 2015;Tan et al., 2015d). SVMPs presumably play a rather minor role in the pathogenesis of cobra envenomation in view of its minute content in the venom.
Vespryn
Thaicobrin is another putative toxin that has been isolated fromN. kaouthiavenom. Even though the protein was structurally characterised two decades ago, its toxic properties and pathogenic role in envenomation have not been fully investigated. The amino acid sequence available from the depository consists of only the mature chain. The present study, however, unmasked the sequences of the signal peptide and pro-peptide region
(Data S3;Fig. 5). Sequencing successfully yielded a full-length vespryn transcript
containing 190 amino acids from both NK-T and NK-M venom glands, with the mature chain being identically matched to the previously reported Thaicobrin (UniProtKB: P82885). Thaicobrin is highly homologous to ohanin isolated fromO. hannahvenom
(Fig. 5), and it is likely that the two proteins exhibit similar pharmacological activities,
both in terms of protein content (Tan et al., 2015d) and gene expression as shown in the present study (∼0.1% of total toxin FPKM).
L-Amino acid oxidase
The present study detected the presence of one and eight LAAO transcripts from the venom glands of NK-M and NK-T, respectively. Among these, NKM_LAO01 and NKT_LAO01 reveal a complete sequence with 514 amino acids length (Table 5). These transcripts comprise the three well-defined domains of LAAO and are highly homologous to LAAO reported from other cobras in particularN. atra(UniProtKB: A8QL58) (Pawelek et al., 2000) (Data S3;Fig. 6). The present study is the first report to reveal the full sequence of N. kaouthiaLAAO (Data S3;Fig. 6), after the previous reported LAAO for N. naja kaouthiaspecimen (unspecified origin, Japan Snake Institute) with solely N-terminal sequence (38 amino acid residues) (Sakurai et al., 2001). Multiple sequence alignment shows the snake venom LAAO sequences appear to be highly conserved across different lineages (Fig. 6) and it is noteworthy that the sequence of the major LAAO from NK-M and NK-T venom glands are completely identical (Data S3andS4). Again, considering that the snake venom LAAO is well conserved and found in the venoms of many lineages, it is most likely evolving under strong purifying selection. LAAO transcripts constitute only about 0.1% of total toxin mRNA, and about 1% of total venom proteins (Tan et al., 2015d). This is another enzyme of low abundance and minimal mutation in most snake venoms, consistent with its rather conserved ancillary function (Du & Clemetson, 2002;Tan et al., 2015e).
Nerve growth factor
[image:24.612.38.580.88.133.2]The present study revealed a NGF transcript (complete sequence with 246 amino acid residues) in the venom glands of both NK-M and NK-T, respectively (Data S2AandS2B). The sequence is fully matched to the ‘nerve growth factor beta chain precursor’ (accession numberA59218, protein information resources – PIR) isolated fromN. kaouthiavenom (Selby, Edwards & Rutter, 1987) (Data S3). This older repository was subsequently replaced by a shorter curated sequence (UniProtKB: P61899) identified from the same species with 116 amino acid residues covering only the mature chain of the NGF. To date, the role of NGF in the toxic action of snake venom remains unconfirmed. However, it has been suggested that the protein may play a role in preventing the autolysis of metalloproteinase auto-digestion as well as to exert cytotoxic and apoptosis-inducing effects (Lavin et al., 2009). Also NGF has also been shown to induce histamine release and may contribute to the hypotensive effect of the venom (Stempelj & Ferjan, 2005).
In the current study, the expression of NGF genes in the venom gland of NK-T and NK-M is low and is in keeping with the minor content of this protein in the venom (<1%) (Tan et al., 2015d).
Other minor toxin transcripts expressed in venom gland
Recent proteomics studies of N. kaouthiavenom revealed the presence of several protein families that exist in low abundance, including KSPI, NP, PDE, 5′NUC and snaclec (Tan et al., 2015d). Results of the currentN. kaouthiatranscriptome confirmed the presence of the above-mentioned protein-encoding genes in both NK-M and NK-T venom glands (Data S2AandS2B;Table 5). All these genes were expressed at a very low level, congruent with their minor composition of the venom proteins.
Two and three KSPI transcripts (all with full sequence) were reported in the venom glands of NK-M and NK-T, respectively. Of these, one gene was co-expressed in NK-M and NK-T, and their sequences were aligned for comparison inData S3. The most abundantly expressed KSPI isoforms in the venom gland of NK-M (NKM_KPI01) and NK-T (NKT_KPI01) is found to be different (Fig. 7). The respective sequences of NK-M and NK-T are highly homologous to serine protease inhibitors reported from other cobra species: N. naja(UniProtKB: P20229) for NK-M transcript, andN. atra(UniProtKB: Q5ZPJ7) for NK-T transcript (Shafqat et al., 1990;Zhou et al., 2004) (Fig. 7).
In this study, partial NP transcripts were also identified and annotated to NPs reported fromN. atra(UniProtKB: D9IX97) (Fig. 8). The venom-derived NP has been shown to induce rapid relaxation of phenylephrine-precontracted rat aortic strips, and to stimulate cGMP production, inducing hypotension in experimental rats (Zhang et al., 2011). It has been reported that most of the NPs found in elapid venoms are of the atrial-NP (ANP), or B-type NP (BNP) which do not have the bradykinin-potentiating peptide (BPP) domain and the part of the linker sequence (Zhang et al., 2011). This is largely consistent with the major NP transcripts found in this study and the ANP and BNP detected at the protein level ofN. kaouthiavenom (Tan et al., 2015d). The present study also detected PDE transcripts in NK-M and NK-T venom glands at very low level of gene expression (<0.01%). Among these, one PDE subtype was co-expressed (also most highly expressed) in both NK-M and NK-T venom glands, and exhibits full-length sequence coverage (NKM_PDE01 and NKT_PDE01) (Data S2AandS2B;Table 5). The two full-length protein-encoding transcripts were identical (Data S3), and are highly homologous to the annotated sequence reported forM. fulvius(UniProtKB: U3FAB3). The pathogenic role of PDE may be shaped towards the potentiation of venom-induced
hypotension and paralysis through the release of purines (Aird, 2002,2005;Dhananjaya & D’Souza, 2010).
Snake venom snaclecs are typical toxins present in viper or pit viper venoms (Arlinghaus & Eble, 2012;Du & Clemetson, 2009). Recent venomic studies showed that snaclecs are not exclusive to viperid venoms—for instance, the presence of snaclecs as a minor protein component has been shown inN. kaouthia venom (Laustsen et al., 2015; Tan et al., 2015d) and this is well supported by their gene expression in this present study. A total of four different snaclecs subtypes were detected in both NK-M and NK-T venom glands, with NKM_SCL01 and NKT_SCL01 being the most highly expressed (<0.3%) (Table 5). The two snaclec transcripts were identical, with sequence homologous to C-type lectins-BFL-1 (UniProtKB: Q90WI8) ofBungarus fasciatus(Data S3). Snaclecs are considered ‘haemotoxic’ toxins for their ability to disrupt platelet functioning (Clemetson, 2010); this effect is however less important in N. kaouthiaenvenoming in this region as thrombocytopaenia attributable to its bite has never been reported.
Novel transcripts detected only at transcriptomic level
[image:26.612.190.579.85.178.2]The transcriptomic study further identified 11 putative toxin families comprising a total of 16 and 16 toxin transcripts, respectively, in the venom gland of NK-M and NK-T. These include cystatin, SVSP, AChE, HYA, CF, PLB, waprin, neprilysin, AP, DPP-IV and IGF. Most of these toxin families have not been reported in cobra venom, presumably due to their very low level of protein expression; while at the transcriptional level, only a
Figure 8 Pairwise sequence alignment of natriuretic peptide (NP) transcripts from the venom gland transcriptomes of NK-M and NK-T in comparison to the annotated NP sequences.Green, consensus sequence; red, sequence diversification; black, mismatched sequences.
[image:26.612.188.580.229.357.2]single cystatin transcript ofN. kaouthia (Thailand) has been reported from an earlier cloning study (Richards et al., 2011). The cystatin transcript, deposited as UniProtKB: E3P6P4, is identical to transcripts NKM_CYS02 and NKT_CYS02 uncovered in this study
(Data S3). Among the four additional cystatin-like transcripts sequenced in the current
study, three of which were co-expressed in NK-M and NK-T (Data S2A,S2BandS3;
Table 5). Of note, NKM_CYS01 and NKT_CYS01 from NK-M and NK-T, respectively,
are the most highly expressed cystatin transcripts and possess novel sequence that is unique from the annotated protein (Data S3). Although the actual role of cystatin remains equivocal, it was suggested that its protease inhibition properties may contribute to the stability of toxin proteins in the venom glands (Richards et al., 2011).
Snake venom serine proteases are generally involved in venom-induced consumptive coagulopathy (Serrano & Maroun, 2005). There is one full-length protein-encoding SVSP transcript detected in both NK-M and NK-T venom glands (transcripts NKM_SSP01 and NKT_SSP01). Both of the transcript from NK-M and NK-T are highly homologous to the SVSP-harobin (UniProtKB: Q5MCS0) from Hydrophis curtus(Fig. 9). However, none of the proteases were detected in the venom proteome (Tan et al., 2015d), indicating that the proteins were either not translated or exist at very low level, consistent with the negative finding of coagulopathy inN. kaouthia envenomation.
[image:27.612.186.580.84.235.2]Acetylcholinesterase (Ahmed et al., 2009) and HYA (Tu & Hendon, 1983) are two well-investigated snake venom enzymes, yet their content in snake venom is usually very low and often undetectable in venom proteome, although the enzymatic activities have been detected in various cobra venoms (Tan & Tan, 1988). In the present study, transcripts of the two enzymes were reported fromN. kaouthiavenom glands, supporting the expression of the proteins in N. kaouthiavenom. Importantly, the full sequences of AChE were unveiled in the venom gland transcriptomes of NK-M (NKM_ACE01) and NK-T (NKT_ACE01), with identical sequences observed (Data S3). This is also the first report of N. kaouthiaAChE sequence (as well as the complete sequence inNaja species), which are homologous to the AChE (UniProtKB: Q92035) reported from B. fasciatus(Data S3). On the other hand, HYA gene expression was detected in NK-T
Figure 9 Pairwise sequence alignment of snake venom serine protease (SVSP) transcripts from the venom gland transcriptomes of NK-M and NK-T in comparison to the annotated SVSP sequences.
venom gland, with full-length protein-encoding transcript (NKT_HYA01) obtained. The HYA transcript (NKT_HYA01) representing the first reported sequences of HYA of N. kaouthia as well as inNajaspecies and share 84% homology with HYA from Echis ocellatus(UniProtKB: A3QVN2) (Fig. 10). HYA gene, a relatively less investigated venom enzyme family, is likely well-conserved across many lineages of Elapidae and Viperidae as a result of purifying selection.
It is interesting to note that the CF X transcript was also detected (albeit at very low abundance) inN. kaouthiavenom gland transcriptomes, even thoughN. kaouthiavenom is not known to exhibit procoagulant activity. Another protein family of the putative toxin is PLB; this is an ‘emerging’ snake venom enzyme with little characterisation, only detected recently in many snake venoms through the high-resolution mass spectrometry technique. Complete sequence for PLB from both NK-M and NK-T venom glands was also obtained in the present study. The transcripts (NKM_PLB01 and NKT_PLB01) with higher expression show sequences that are highly homologous to PLB-81b isolated fromDrysdalia coronoides(Chatrath et al., 2011) (Data S3). This is also the first report of PLB sequence inNajaspecies though the activity has been reported inN. najavenom (Shiloah, 1974).
[image:28.612.188.577.85.239.2]Waprins with little known toxic properties have not been extensively reported. The current study first reported the detection of waprins at the transcriptional level in Najaspecies (one transcripts in NK-M and NK-T venom glands, FPKM < 0.01%). The transcripts, with full amino acid sequences, were annotated to Scuwaprin-a (Oxyuranus scutellatus scutellatus) (Data S3;Fig. 11). Neprilysin-like protein is a novel metalloendopeptidase that has a wide range of functional targets in the regulatory processes of natriuretic and vasodilatory neuropeptides (Turner, Isaac & Coates, 2001). It has been reported as putative toxins in the saw-scaled viper and king cobra (Casewell et al., 2009;Tan et al., 2015a;Vonk et al., 2013), though its role in the pathogenesis of envenomation has not been established. In this study, neprilysin-like transcripts were detected in both venom glands. The transcripts are identical between the both
samples, while the one from NK-T shows a full sequence. This represents a novel full amino acid sequence of neprilysin-like protein in Najaspecies, which shares a high degree of homology to the neprilysin identified from Python bivittatus(Data S3;
Table 5). The finding may reflect the presence of venom gene orthologues in the
ancestral genome prior to the expansion and diversification of various snake lineages (Reyes-Velasco et al., 2015).
The present study also showed the presence of several transcripts encoding APs (two subtypes), dipeptidylpepidase-IV (one transcript each sample, with full sequence) and IGFs (one transcript each sample, with full sequences). These can at best be considered as putative toxins as the functionalities of these proteins have not been rigorously established. APs are exo-metalloproteases that function in the physiological maintenance of the blood pressure (Vaiyapuri et al., 2010), while DPP-IV is a highly glycosylated serine protease that may counteract the hypertensive response in the envenomed prey by destroying hypertensive peptidyl hormones (Aird, 2008). The level of expression of these three putative toxins was very low (<0.03%) and the expressed proteins were not detectable even with the use of highly sensitive nano-LCMS/MS technique (Tan et al., 2015d), although some authors reported the presence of AP protein in certain snake venoms (Faiz et al., 1996;Gasparello-Clemente & Silveira, 2002). ForN. kaouthia, full sequences of DPP-IV and IGF were available from the present study and these are the only ones reported from the venom gland transcriptomes ofNajaspecies thus far.
CONCLUSION
This study set to elucidate the venom gland transcriptomes of N. kaouthiafrom two different geographical origins: Malaysia and Thailand. The findings demonstrated the unique expression patterns of toxin-encoding gene transcripts with high redundancy in comparison to the non-toxin genes. A total of 22 venom gene families were
[image:29.612.189.575.87.187.2]co-expressed in both NK-M and NK-T venom glands (out of the 24 families identified), with 15 gene families reported for the first time inN. kaouthiaat transcriptional level. Comparatively, the expressions of venom genes in NK-M and NK-T were generally comparable while being dominated by 3FTxs. Inter-locale variations were remarkable in the magnitude of gene expression, for example, NK-T transcriptome is dominated by LNTX expression, whereas NK-M is dominated by SNTX; CTX-3 was exclusively expressed in NK-T, whereas CTX-5 is the most abundantly expressed CTX gene in