Acta Cryst.(2002). E58, o999±o1000 DOI: 10.1107/S160053680201423X Peter G. Joneset al. C9H11F2P
o999
organic papers
Acta Crystallographica Section E Structure Reports
Online
ISSN 1600-5368
Difluoro(mesityl)phosphine
Peter G. Jones,* Lutz Heuer and Reinhard Schmutzler
Institut fuÈr Anorganische und Analytische Chemie, Technische UniversitaÈt Braunschweig, Postfach 3329, 38023 Braunschweig, Germany
Correspondence e-mail: [email protected]
Key indicators Single-crystal X-ray study
T= 178 K
Mean(C±C) = 0.002 AÊ Disorder in main residue
Rfactor = 0.030
wRfactor = 0.087
Data-to-parameter ratio = 14.6
For details of how these key indicators were automatically derived from the article, see http://journals.iucr.org/e.
#2002 International Union of Crystallography Printed in Great Britain ± all rights reserved
The title compound, C9H11F2P, displays mirror symmetry, with all ring C and the P atom lying in the mirror plane. The methyl H atoms are disordered across the mirror plane. Key dimensions around the P atom are PÐF 1.5814 (10), PÐC 1.8116 (17) AÊ and FÐPÐF 95.95 (8).
Comment
We are interested in organic derivatives of phosphorus tri¯uoride. We have published the syntheses of several compounds of the type RPF2 (Heuer & Schmutzler, 1988, 1989; Wesemannet al., 1992, and references therein), but the X-ray structure determinations have, with one exception (Heuer et al., 1989), been limited to metal complexes. We present here the structure of uncomplexed di¯uoro(mesityl)-phosphine, (I).
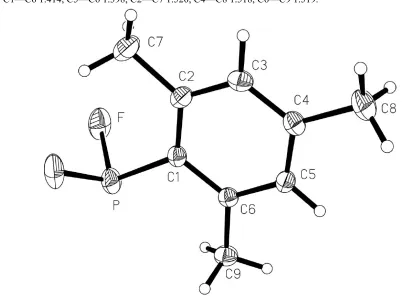
The molecule is shown in Fig. 1; it possesses crystallographic mirror symmetry, with all atoms except ¯uorine and methyl H atoms (which are disordered, see Experimental) lying in the mirror planes. A closely similar arrangement was observed for the hydrogen analogue mesitylphosphine (Bartlett et al., 1987), which also crystallizes inPnma, but the structures do not seem to be isotypic.
The molecular dimensions of (I) may be regarded as normal (Table 1). For discussion purposes, non-corrected bond lengths
Received 1 August 2002 Accepted 7 August 2002 Online 16 August 2002
Figure 1
are used, but libration-corrected values are presented in the Experimentalsection. The PÐF bond length is 1.5814 (10) AÊ, with FÐPÐF 95.95 (8),cf. 1.572, 1.581 (2) AÊ and 96.3 (1)in the anthracene-9-PF2 dimer (Heuer et al., 1989). The PÐC bond length of 1.8116 (17) AÊ may be compared with the value of 1.807 (5) AÊ observed for mesitylphosphine (Bartlettet al., 1987), but is rather shorter than the average of 1.833 AÊ observed for the series PPh2(Mes), PPh(Mes)2 and P(Mes)3 (Mes is mesityl; Blountet al., 1994), and much shorter than the 1.859 (2) AÊ in the anthracene dimer, for which steric crowding may be responsible for bond lengthening.
The orientation of the PF2group with respect to the ring is given by the torsion angle FÐPÐC1ÐC2 of 49.15 (4).
One of the methyl H atoms is involved in a short contact to the centroid (Cg) of a ring in the neighbouring layer: C9Ð H9A Cg with normalized H Cg 2.64 AÊ and CÐH Cg 150. This could be considered as a CÐH interaction.
Experimental
The title compound was prepared from the reaction of the cor-responding chloride with sodium ¯uoride in acetonitrile. It was isolated as an oil that crystallized in the form of colourless prisms (m.p. 408±410 K), extremely sensitive to moist air (Heuer, 1989). For this reason, the crystal used for structure determination was rather larger than usual.
Crystal data
C9H11F2P Mr= 188.15 Orthorhombic,Pnma a= 13.261 (5) AÊ
b= 7.185 (2) AÊ
c= 9.735 (3) AÊ
V= 927.6 (5) AÊ3 Z= 4
Dx= 1.347 Mg mÿ3
MoKradiation Cell parameters from 50
re¯ections
= 8±11.5 = 0.27 mmÿ1 T= 178 (2) K Block, colourless 0.70.70.7 mm
Data collection
NicoletR3 diffractometer
!scans
2236 measured re¯ections 1154 independent re¯ections 1014 re¯ections withI> 2(I)
Rint= 0.015 max= 27.5
h= 0!17
k= 0!9
l=ÿ12!12 3 standard re¯ections
every 147 re¯ections intensity decay: 1.5%
Re®nement
Re®nement onF2 R[F2> 2(F2)] = 0.030 wR(F2) = 0.087 S= 1.09 1154 re¯ections 79 parameters
H atoms treated by a mixture of independent and constrained re®nement
w= 1/[2(F
o2) + (0.0397P)2 + 0.2323P]
whereP= (Fo2+ 2Fc2)/3 (/)max= 0.001
max= 0.21 e AÊÿ3
min=ÿ0.34 e AÊÿ3
Table 1
Selected geometric parameters (AÊ,).
PÐF 1.5814 (10) PÐC1 1.8116 (17)
FÐPÐFi 95.95 (8) FÐPÐC1 100.88 (5)
FÐPÐC1ÐC2 49.15 (4) Symmetry code: (i)x;1
2ÿy;z.
The aromatic atoms H3 and H5 were re®ned freely. The methyl H atoms, which are disordered across the mirror plane, could none-theless be identi®ed in difference syntheses; the methyl groups were then idealized and re®ned as rigid groups allowed to rotate but not tip. The structure could also be re®ned with ordered methyl groups in the corresponding non-centrosymmetric space groupPna21, but with
a series of restraints to improve re®nement stability. TheR values were not improved. It is unlikely that X-ray methods alone can distinguish between the two possibilities; we prefer the disordered centrosymmetric model because it is unaffected by the matrix near-singularities inherent in thePna21re®nement. The latter is presented
in the deposited material. A rigid-body libration correction (Scho-maker & Trueblood, 1968) gave the following corrected bond lengths (AÊ): PÐF 1.588, PÐC1 1.819, C1ÐC2 1.419, C2ÐC3 1.397, C3ÐC4 1.392, C4ÐC5 1.391, C1ÐC6 1.414, C5ÐC6 1.398, C2ÐC7 1.520, C4ÐC8 1.518, C6ÐC9 1.519.
Data collection: P3 (Nicolet, 1987); cell re®nement: P3; data reduction:XDISK(Nicolet, 1987); program(s) used to solve struc-ture:SHELXS97 (Sheldrick, 1990); program(s) used to re®ne struc-ture: SHELXL97 (Sheldrick, 1997); molecular graphics: XP
(Siemens, 1994); software used to prepare material for publication:
SHELXL97.
Financial support from the Fonds der Chemischen Industrie is gratefully acknowledged. The authors thank Mr A. Wein-kauf for technical assistance.
References
Bartlett, R. A., Olmstead, M. M., Power, P. P. & Sigel, G. A. (1987).Inorg. Chem.26, 1941±1946.
Blount, J. F., Camp, D., Hart, R. D., Healy, P. C., Skelton, B. W. & White, A. H. (1994).Aust. J. Chem.47, 1631±1639.
Heuer, L. (1989). PhD dissertation, Technical University of Braunschweig, Germany.
Heuer, L. & Schmutzler, R. (1988).J. Fluorine Chem.39, 197±216. Heuer, L. & Schmutzler, R. (1989).J. Fluorine Chem.45, 32.
Heuer, L., Schomburg, D. & Schmutzler, R. (1989).Chem. Ber.122, 1473± 1476.
Nicolet (1987).P3 andXDISK.Nicolet Instrument Corporation, Madison, Wisconsin, USA.
Schomaker, V. & Trueblood, K. N. (1968).Acta Cryst.B24, 63±76. Sheldrick, G. M. (1990).Acta Cryst.A46, 467±473.
Sheldrick, G. M. (1997).SHELXL97. University of GoÈttingen, Germany. Siemens (1994).XP. Version 5.03. Siemens Analytical X-ray Instruments Inc.,
Madison, Wisconsin, USA.
Wesemann, J., Jones, P. G., Schomburg, D., Heuer, L. & Schmutzler, R. (1992).
supporting information
sup-1
Acta Cryst. (2002). E58, o999–o1000supporting information
Acta Cryst. (2002). E58, o999–o1000 [doi:10.1107/S160053680201423X]
Difluoro(mesityl)phosphine
Peter G. Jones, Lutz Heuer and Reinhard Schmutzler
S1. Comment
We are interested in organic derivatives of phosphorus trifluoride. We have published the syntheses of several compounds
of the type RPF2 (Heuer & Schmutzler, 1988, 1989; Wesemann et al., 1992, and references therein), but the X-ray
structure determinations have, with one exception (Heuer et al., 1989), been limited to metal complexes. We present here
the structure of uncomplexed difluoro(mesityl)phosphine, (I).
The molecule is shown in Fig. 1; it possesses crystallographic mirror symmetry, with all atoms except fluorine and
methyl H atoms (which are disordered, see Experimental) lying in the mirror planes. A closely similar arrangement was
observed for the hydrogen analogue mesitylphosphine (Bartlett et al., 1987), which also crystallizes in Pnma, but the
structures do not seem to be isotypic.
The molecular dimensions of (I) may be regarded as normal. For discussion purposes, non-corrected bond lengths are
used, but libration-corrected values are presented in the Experimental section. The P—F bond length is 1.5814 (10) Å,
with F—P—F 95.95 (8)°, cf. 1.572, 1.581 (2) Å and 96.3 (1)° in the anthracene-9-PF2 dimer (Heuer et al., 1989). The P—
C bond length of 1.8116 (17) Å may be compared with the value of 1.807 (5) Å observed for mesitylphosphine (Bartlett
et al., 1987), but is rather shorter than the average of 1.833 Å observed for the series PPh2(Mes), PPh(Mes)2 and P(Mes)3
(Blount et al., 1994), and much shorter than the 1.859 (2) Å in the anthracene dimer, for which steric crowding may be
responsible for bond lengthening.
The orientation of the PF2 group with respect to the ring is given by the torsion angle F—P—C1—C2 of 49.15 (4)°.
One of the methyl H atoms is involved in a short contact to the centroid (Cg) of a ring in the neighbouring layer: C9—
H9A···Cg with normalized H···Cg 2.64 Å and C—H···Cg 150°. This could be considered as a C—H···π interaction.
S2. Experimental
The title compound was prepared from the reaction of the corresponding chloride with sodium fluoride in acetonitrile. It
was isolated as an oil that crystallized in the form of colourless prisms (m.p. 408–410 K), extremely sensitive to
laboratory air (Heuer, 1989). For this reason, the crystal used for structure determination was rather larger than usual.
S3. Refinement
The aromatic atoms H3 and H5 were refined freely. The methyl H atoms, which are disordered across the mirror plane,
could nonetheless be identified in difference syntheses; the methyl groups were then idealized and refined as rigid groups
allowed to rotate but not tip. The structure could also be refined with ordered methyl groups in the corresponding
non-centrosymmetric space group Pna21, but with a series of restraints to improve refinement stability. The R values were not
improved. It is unlikely that X-ray methods alone can distinguish between the two possibilities; we prefer the disordered
centrosymmetric model because it is unaffected by the matrix near-singularities inherent in the Pna21 refinement. The
following corrected bond lengths (Å): P—F 1.588, P—C1 1.819, C1—C2 1.419, C2—C3 1.397, C3—C4 1.392, C4—C5
[image:4.610.85.505.95.397.2]1.391, C1—C6 1.414, C5—C6 1.398, C2—C7 1.520, C4—C8 1.518, C6—C9 1.519.
Figure 1
The molecule of the title compound in the crystal. Displacement ellipsoids are shown at the 30% probability level.
H-atom radii are arbitrary.
Mesityldifluorophosphine
Crystal data
C9H11F2P Mr = 188.15
Orthorhombic, Pnma a = 13.261 (5) Å b = 7.185 (2) Å c = 9.735 (3) Å V = 927.6 (5) Å3 Z = 4
F(000) = 392
Dx = 1.347 Mg m−3
Mo Kα radiation, λ = 0.71073 Å Cell parameters from 50 reflections θ = 8–11.5°
µ = 0.27 mm−1 T = 178 K Block, colourless 0.7 × 0.7 × 0.7 mm
Data collection
Nicolet R3 diffractometer
Radiation source: fine-focus sealed tube Graphite monochromator
ω scans
2236 measured reflections 1154 independent reflections 1014 reflections with I > 2σ(I)
Rint = 0.015
θmax = 27.5°, θmin = 3.1° h = 0→17
k = 0→9 l = −12→12
supporting information
sup-3
Acta Cryst. (2002). E58, o999–o1000Refinement
Refinement on F2 Least-squares matrix: full R[F2 > 2σ(F2)] = 0.030 wR(F2) = 0.087 S = 1.09 1154 reflections 79 parameters 0 restraints
Primary atom site location: structure-invariant direct methods
Secondary atom site location: difference Fourier map
Hydrogen site location: inferred from neighbouring sites
H atoms treated by a mixture of independent and constrained refinement
w = 1/[σ2(F
o2) + (0.0397P)2 + 0.2323P] where P = (Fo2 + 2Fc2)/3
(Δ/σ)max = 0.001 Δρmax = 0.21 e Å−3 Δρmin = −0.34 e Å−3
Special details
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes.
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger.
Final refinement of the alternative model in Pna2(1):
TITL MESFO in Pna21 CELL 0.71073 13.261 9.735 7.185 90.00 90.00 90.00 ZERR 4.00 0.005 0.003 0.002 0.00 0.00 0.00 L A T T −1 SYMM −x,-y,0.5 + z SYMM 0.5 + x,0.5 − y,z SYMM 0.5 − x,0.5 + y,0.5 + z SFAC C H F P UNIT 36 44 8 4 TEMP −95 SIZE 0.7 0.7 0.7 L·S. 4 FMAP 2 PLAN 10 DELU SIMU SADI C7 H7A C7 H7B C7 H7C C8 H8A C8 H8B C8 H8C C9 H9A C9 H9B C9 H9C SADI H7A H7B H7A H7C H7B H7C H8A H8B H8A H8C H8B H8C H9A H9B H9A H9C H9B H9C WGHT 0.047300 0.202000 FVAR 2.32532 P 4 0.423192 0.744056 0.520125 11.00000 0.06015 0.02825 = 0.05304 − 0.00086 0.00024 0.00593 F1 3 0.349251 0.785341 0.683751 11.00000 0.09525 0.03992 = 0.05375 − 0.00739 0.01003 0.02203 F2 3 0.347575 0.779485 0.356626 11.00000 0.09522 0.05638 = 0.04619 0.01154 0.00099 0.03171 flat c1 > c6 same c1 c6 < c2 C1 1 0.412749 0.558693 0.521719 11.00000 0.03680 0.02923 = 0.02905 0.00214 0.00366 0.00289 C2 1 0.504768 0.486457 0.519958 11.00000 0.03191 0.03253 = 0.02798 − 0.00180 0.00149 − 0.00040 C3 1 0.502870 0.343557 0.519671 11.00000 0.03574 0.03256 = 0.03439 0.00359 0.00461 0.00608 AFIX 43 H3 2 0.564896 0.294732 0.519124 11.00000 − 1.20000 AFIX 0 C4 1 0.413397 0.270347 0.520157 11.00000 0.04622 0.03081 = 0.03533 − 0.00062 0.00087 − 0.00256 C5 1 0.323307 0.342865 0.521671 11.00000 0.03544 0.04239 = 0.03943 0.00058 0.00236 − 0.00813 AFIX 43 H5 2 0.261537 0.293449 0.523036 11.00000 − 1.20000 AFIX 0 C6 1 0.320696 0.485580 0.521251 11.00000 0.03199 0.04281 = 0.03397 − 0.00144 − 0.00119 0.00475 C7 1 0.606354 0.556836 0.522338 11.00000 0.03518 0.04656 = 0.04453 0.00333 0.00650 − 0.00637 H7A 2 0.658118 0.487797 0.563976 11.00000 0.04088 H7B 2 0.611256 0.635593 0.612886 11.00000 0.04457 H7C 2 0.626626 0.589962 0.396627 11.00000 0.06572 C8 1 0.415147 0.115216 0.520921 11.00000 0.07173 0.03077 = 0.06755 0.00571 0.00545 − 0.00478 H8A 2 0.423830 0.080404 0.391482 11.00000 0.07896 H8B 2 0.349790 0.080626 0.571594 11.00000 0.06340 H8C 2 0.471017 0.076989 0.599913 11.00000 0.05253 C9 1 0.218608 0.554852 0.522924 11.00000 0.03409 0.06773 = 0.06689 0.00526 − 0.00348 0.01188 H9A 2 0.167635 0.488153 0.568060 11.00000 0.05191 H9B 2 0.200722 0.583079 0.392721 11.00000 0.07149 H9C 2 0.217295 0.638548 0.602643 11.00000 0.04823 HKLF 4 1 1 0 0 0 0 1 0 − 1 0
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
x y z Uiso*/Ueq Occ. (<1)
H3 0.2582 (17) 0.2500 0.296 (2) 0.060 (6)* C4 0.41334 (13) 0.2500 0.27039 (17) 0.0376 (4) C5 0.50289 (12) 0.2500 0.34348 (16) 0.0341 (3) H5 0.5663 (15) 0.2500 0.298 (2) 0.047 (5)* C6 0.50481 (12) 0.2500 0.48651 (16) 0.0308 (3) C7 0.21840 (14) 0.2500 0.5549 (2) 0.0562 (5)
H7A 0.2206 0.3300 0.6363 0.067* 0.50 H7C 0.2009 0.1228 0.5823 0.067* 0.50 H7B 0.1675 0.2972 0.4907 0.067* 0.50 C8 0.41505 (17) 0.2500 0.1151 (2) 0.0564 (5)
H8A 0.3511 0.2996 0.0802 0.068* 0.50 H8B 0.4243 0.1225 0.0817 0.068* 0.50 H8C 0.4708 0.3280 0.0826 0.068* 0.50 C9 0.60655 (13) 0.2500 0.55665 (19) 0.0421 (4)
H9A 0.6224 0.3761 0.5884 0.051* 0.50 H9B 0.6583 0.2088 0.4916 0.051* 0.50 H9C 0.6049 0.1651 0.6354 0.051* 0.50
Atomic displacement parameters (Å2)
U11 U22 U33 U12 U13 U23
P 0.0602 (3) 0.0532 (3) 0.0281 (2) 0.000 0.00585 (19) 0.000 F 0.0953 (7) 0.0497 (5) 0.0486 (5) −0.0044 (5) 0.0269 (4) 0.0092 (4) C1 0.0372 (8) 0.0288 (7) 0.0295 (7) 0.000 0.0029 (6) 0.000 C2 0.0319 (8) 0.0335 (8) 0.0434 (9) 0.000 0.0048 (6) 0.000 C3 0.0354 (8) 0.0392 (9) 0.0430 (9) 0.000 −0.0082 (7) 0.000 C4 0.0467 (9) 0.0350 (8) 0.0309 (8) 0.000 −0.0025 (6) 0.000 C5 0.0352 (8) 0.0340 (8) 0.0331 (8) 0.000 0.0060 (6) 0.000 C6 0.0318 (7) 0.0274 (7) 0.0331 (7) 0.000 −0.0002 (5) 0.000 C7 0.0348 (9) 0.0662 (13) 0.0678 (13) 0.000 0.0117 (9) 0.000 C8 0.0713 (13) 0.0667 (13) 0.0311 (9) 0.000 −0.0056 (8) 0.000 C9 0.0360 (8) 0.0442 (9) 0.0461 (9) 0.000 −0.0057 (7) 0.000
Geometric parameters (Å, º)
supporting information
sup-5
Acta Cryst. (2002). E58, o999–o1000F—P—Fi 95.95 (8) C1—C6—C9 123.29 (14) F—P—C1 100.88 (5) C2—C7—H7A 109.5 Fi—P—C1 100.88 (5) C2—C7—H7C 109.5 C6—C1—C2 119.98 (14) H7A—C7—H7C 109.5 C6—C1—P 115.48 (12) C2—C7—H7B 109.5 C2—C1—P 124.54 (12) H7A—C7—H7B 109.5 C3—C2—C1 118.70 (14) H7C—C7—H7B 109.5 C3—C2—C7 117.86 (16) C4—C8—H8A 109.5 C1—C2—C7 123.44 (16) C4—C8—H8B 109.5 C4—C3—C2 122.01 (15) H8A—C8—H8B 109.5 C4—C3—H3 121.6 (14) C4—C8—H8C 109.5 C2—C3—H3 116.4 (14) H8A—C8—H8C 109.5 C5—C4—C3 118.51 (15) H8B—C8—H8C 109.5 C5—C4—C8 120.06 (16) C6—C9—H9A 109.5 C3—C4—C8 121.43 (16) C6—C9—H9B 109.5 C4—C5—C6 121.97 (15) H9A—C9—H9B 109.5 C4—C5—H5 121.3 (13) C6—C9—H9C 109.5 C6—C5—H5 116.7 (13) H9A—C9—H9C 109.5 C5—C6—C1 118.82 (14) H9B—C9—H9C 109.5 C5—C6—C9 117.89 (14)
F—P—C1—C6 −130.85 (4) C2—C3—C4—C5 0.0 Fi—P—C1—C6 130.85 (4) C2—C3—C4—C8 180.0 F—P—C1—C2 49.15 (4) C3—C4—C5—C6 0.0 Fi—P—C1—C2 −49.15 (4) C8—C4—C5—C6 180.0 C6—C1—C2—C3 0.0 C4—C5—C6—C1 0.0 P—C1—C2—C3 180.0 C4—C5—C6—C9 180.0 C6—C1—C2—C7 180.0 C2—C1—C6—C5 0.0 P—C1—C2—C7 0.0 P—C1—C6—C5 180.0 C1—C2—C3—C4 0.0 C2—C1—C6—C9 180.0 C7—C2—C3—C4 180.0 P—C1—C6—C9 0.0