Acta Cryst.(2001). E57, o63±o65 DOI: 101107/S1600536800019097 William Clegget al. C14H11BO2
o63
organic papers
Acta Crystallographica Section E Structure Reports
Online
ISSN 1600-5368
A 2-styrylboronate ester
William Clegg,a* Todd B.
Marder,b,cAndrew J. Scott,a
Christian Wiesauerc,dand Walter
Weissensteinerd
aDepartment of Chemistry, University of
Newcastle upon Tyne, Newcastle upon Tyne NE1 7RU, England,bDepartment of Chemistry,
University of Durham, South Road, Durham DH1 3LE, England,cDepartment of Chemistry,
University of Waterloo, Waterloo, Ontario, Canada N2L 3G1, anddInstitute of Organic
Chemistry, University of Vienna, Waehringer-strasse 38, A-1090 Wien, Austria
Correspondence e-mail: w. [email protected]
Key indicators
Single-crystal X-ray study
T= 160 K
Mean(C±C) = 0.002 AÊ
Rfactor = 0.037
wRfactor = 0.095
Data-to-parameter ratio = 16.6
For details of how these key indicators were automatically derived from the article, see http://journals.iucr.org/e.
#2001 International Union of Crystallography Printed in Great Britain ± all rights reserved
Molecules of the title compound, (E)-2-(2-phenylethenyl)-1,3,2-benzodioxaborole, C14H11BO2, are essentially
comple-tely planar with a high degree of conjugation. They pack with a herring-bone pattern in the crystal structure. The compound is synthesized with high regioselectivity by hydroboration of phenylacetylene with catecholborane.
Comment
Alkenylboronic esters and acids are versatile synthetic inter-mediates (Lane & Kabalka, 1976) for products such as cis-alkenes and halocis-alkenes, and they have important applica-tions in palladium-catalysed cross-coupling reacapplica-tions and the formation of organometallic compounds (Suzuki, 1985, 1998; Oheet al., 1988; Larocket al., 1972; Pappo & Collins, 1972). The title compound, (I), was obtained by a hydroboration reaction of phenylacetylene with catecholborane (Brown & Gupta, 1972, 1975; Brown & Chandrasekharan, 1983). The ®rst reported preparation of this compound (Joyet al., 1966) involved treatment of cyclooctatetraene with boron trichloride, followed by reaction of the resulting (E)-styryl-boron dichloride with catechol. In order to study the in¯uence of electron-donating and electron-withdrawing substituents at the para-position of the phenyl ring on the reactivity of the double bond in catalysed hydroborations of styrylboronate esters, a series of para-substituted analogues has been prepared (Wiesauer, 1997).
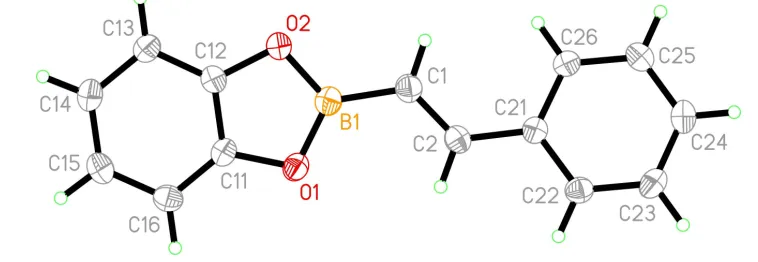
The molecule (Fig. 1) is essentially completely planar, with an r.m.s. deviation of 0.071 AÊ from the least-squares plane through all atoms. All torsion angles are close to 0 and 180,
the largest corresponding to a twist of about 6about the C2Ð
C21 bond linking the phenyl group to the central alkene unit. Bond lengths and angles are normal, with BÐC somewhat shorter and C C longer than generally observed for single and double bonds, respectively (Allen et al., 1992); together with the planarity of the molecule, they indicate extensive delocalization.
It is instructive to compare the title compound with two closely related styryl bis(boronate) esters, (Z)-[(4-NC±C6H4)±
C(Bcat) CH(Bcat)] [(II); Bcat is 1,3,2-benzodioxaborole; cat = 1,2-O2C6H4; Clegget al., 1996] and (Z)-[(4-MeO±C6H4)±
organic papers
o64
William Clegget al. C14H11BO2 Acta Cryst.(2001). E57, o63±o65C(Bcat) CH(Bcat)] [(III); Lesley et al., 1996]. These differ from (I) in having either a-acceptor or-donor substituent at the para-position of the phenyl ring attached to the -carbon of the alkene and also a second Bcat moiety at C. In
(II) and (III), the Bcat at the -position is approximately coplanar (dihedral angles approximately 9 and 7,
respec-tively) with the alkene unit, as in (I), whereas the Bcat groups at Cin (II) and (III) are roughly perpendicular (80 and 84,
respectively) to the alkene plane. In both (II) and (III), the CÐB distances [1.535 (2) and 1.526 (2) AÊ] are signi®cantly
shorter than their CÐB distances [1.569 (2) and 1.566 (2) AÊ]
and are consistent with the CÐB distance in (I) of
1.5321 (17) AÊ. Presumably this is a re¯ection of the delocali-zation present when the Bcat group is coplanar with the alkene andtransto the aryl moiety. The C C distance in (I) [1.3368 (16) AÊ] is shorter than the corresponding distance in either (II) or (III) [1.349 (2) and 1.348 (2) AÊ, respectively] and is probably a result of less steric repulsion in the less substi-tuted alkene.
This structure was recently used as a test of crystal structure prediction methods (Lommerseet al., 2000), and proved to be a considerable challenge to the currently available software systems, with only one prediction reasonably close to the experimentally observed structure.
Experimental
In a nitrogen-®lled glove-box, phenylacetylene (4.0 g, 39 mmol) and catecholborane (4.7 g, 39 mmol) were heated at 343 K for 2 h in a scintillation vial. On cooling, an orange solid was formed. The pure product was obtained as a colourless crystalline material by recrys-tallization from diethylether/hexane by solvent diffusion at 238 K, in 70% yield. Analysis calculated: C 75.73, H 4.99%; found: C 75.30, H 4.98%.1H NMR (200 MHz):6.48 (d, J= 18.5 Hz, 1H, CH CHB),
7.08 (m, 2H, C6H4), 7.26 (m, 2H, C6H4), 7.38 (m, 3H, C6H5), 7.58 (m,
2H, C6H5), 7.77 (d, J = 18.5 Hz, 1H, CH CHB). 13C{1H} NMR
(50 MHz):112.3 (2C, C3and C6of C6H4), 122.6 (2C, C4and C5of
C6H4), 127.4 (2C, Ph), 128.7 (2C, Ph), 129.6 (1C, C4of Ph), 136.9 (1C,
C1of Ph), 148.3 (2C, C1and C2of C6H4), 152.0 (1C,CH CHB),
resonances of C bonded to B too broad to be observed.11B{1H} NMR
(64 MHz):31.3. MS: 222 (M+, 100), 207 (26), 196 (52), 179 (30), 178
(30), 144 (68), 136 (78), 129 (21), 120 (84), 111 (60), 110 (79), 102 (77), 92 (48%).
Crystal data
C14H11BO2 Mr= 222.04
Monoclinic,P21/c a= 6.8351 (9) AÊ b= 7.6342 (10) AÊ c= 21.422 (3) AÊ
= 96.447 (3) V= 1110.8 (3) AÊ3 Z= 4
Dx= 1.328 Mg mÿ3
MoKradiation Cell parameters from 5221
re¯ections
= 2.8±28.6 = 0.09 mmÿ1 T= 160 (2) K Block, colourless 0.620.500.40 mm
Data collection
Bruker SMART 1K CCD diffract-ometer
!rotation with narrow frames 6709 measured re¯ections 2571 independent re¯ections 2326 re¯ections withI> 2(I)
Rint= 0.022 max= 28.6 h=ÿ9!8 k=ÿ6!10 l=ÿ28!28
Re®nement
Re®nement onF2 R[F2> 2(F2)] = 0.037 wR(F2) = 0.095 S= 1.06 2571 re¯ections 155 parameters
H-atom parameters constrained
w= 1/[2(F
o2) + (0.0389P)2
+ 0.3966P]
whereP= (Fo2+ 2Fc2)/3
(/)max= 0.001 max= 0.26 e AÊÿ3 min=ÿ0.15 e AÊÿ3
Extinction correction:SHELXTL Extinction coef®cient: 0.021 (2)
Table 1
Selected geometric parameters (AÊ,).
C1ÐC2 1.3368 (16)
C1ÐB1 1.5321 (17)
C2ÐC21 1.4692 (15)
B1ÐO2 1.3909 (15)
B1ÐO1 1.3910 (15)
C2ÐC1ÐB1 124.72 (11)
C1ÐC2ÐC21 126.86 (11)
O2ÐB1ÐO1 111.54 (10)
O2ÐB1ÐC1 122.96 (11)
O1ÐB1ÐC1 125.50 (11)
C11ÐO1ÐB1 104.82 (9)
C12ÐO2ÐB1 105.06 (9)
B1ÐC1ÐC2ÐC21 ÿ178.83 (11) C2ÐC1ÐB1ÐO2 179.17 (11) C2ÐC1ÐB1ÐO1 ÿ1.22 (19)
C1ÐC2ÐC21ÐC22 173.52 (11) C1ÐC2ÐC21ÐC26 ÿ6.08 (18)
The data collection nominally covered over a hemisphere of reciprocal space, by a combination of three sets of exposures; each set had a different'angle for the crystal and each exposure covered 0.3
in !. H atoms were placed geometrically and re®ned with a riding model (including free rotation about CÐC bonds), and with Uiso
constrained to be 1.2Ueqof the carrier atom.
Data collection: SMART(Siemens, 1995); cell re®nement: local programs; data reduction:SAINT(Siemens, 1995); program(s) used to solve and re®ne structure:SHELXTL(Sheldrick, 1994); molecular graphics:SHELXTL; software used to prepare material for publi-cation:SHELXTLand local programs.
WC and AJS thank EPSRC (UK) for ®nancial support. TBM thanks NSERC of Canada for research support, and the University of Newcastle upon Tyne for a Visiting Senior Research Fellowship. TBM and WC thank NSERC and The Royal Society for support of this collaboration through the Bilateral Exchange Program. Financial support to WW by Fonds zur FoÈrderung der wissenschaftlichen Forschung
Figure 1
(project 10474-CHE) is fratefully acknowledged. CW thanks the Bundesministerium fuÈr Wissenschaft und Verkehr for supporting his six-month stay at the University of Waterloo.
References
Allen, F. H., Kennard, O., Watson, D. G., Brammer, L., Orpen, A. G. & Taylor, R. (1992).International Tables for Crystallography, Vol. C, pp. 685±706. Dordrecht: Kluwer Academic Publishers.
Brown, H. C. & Gupta, S. K. (1972).J. Am. Chem. Soc.94, 4370±4371. Brown, H. C. & Gupta, S. K. (1975).J. Am. Chem. Soc.97, 5249±5255. Brown, H. C. & Chandrasekharan, J. (1983). J. Org. Chem. 48, 5080±
5082.
Clegg, W., Scott, A. J., Lesley, G., Marder, T. B. & Norman, N. C. (1996).Acta Cryst.C52, 1991±1995.
Joy, F., Lappert, M. F. & Prokai, B. (1966).J. Organomet. Chem.5, 506± 519.
Lane, C. F. & Kabalka, G. W. (1976).Tetrahedron,32, 981±990.
Larock, R. C., Gupta, S. K. & Brown, H. C. (1972).J. Am. Chem. Soc.94, 4371± 4373.
Lesley, G., Nguyen, P., Taylor, N. J., Marder, T. B., Scott, A. J., Clegg, W. & Norman, N. C. (1996).Organometallics,15, 5137±5154.
Lommerse, J. P. M., Motherwell, W. D. S., Ammon, H. L., Dunitz, J. D., Gavezzotti, A., Hofmann, D. W. M., Leusen, F. J. J., Mooij, W. T. M., Price, S. L., Schweizer, B., Schmidt, M. U., van Eijck, B. P., Verwer, P. & Williams, D. E. (2000).Acta Cryst.B56, 697±714.
Ohe, T., Ohe, K., Uemura, S. & Sugita, N. (1988).J. Organomet. Chem.344, C5±7.
Pappo, R. & Collins, P. W. (1972).Tetrahedron Lett.pp. 2627±2630. Sheldrick, G. M. (1994).SHELXTL.Version 5. Siemens Analytical X-ray
Instruments Inc., Madison, Wisconsin, USA.
Siemens (1995).SMARTandSAINT. Siemens Analytical X-ray Instruments Inc., Madison, Wisconsin, USA.
Suzuki, A. (1985).Pure Appl. Chem.57, 1749±1758.
Suzuki, A. (1998).Metal-Catalyzed Cross-Coupling Reactions, edited by F. Diederich & P. J. Stang, pp. 49±97. Weinheim: Wiley-VCH.
Wiesauer, C. (1997). Doctoral thesis, University of Vienna, Austria.
Acta Cryst.(2001). E57, o63±o65 William Clegget al. C14H11BO2
o65
supporting information
sup-1
Acta Cryst. (2001). E57, o63–o65
supporting information
Acta Cryst. (2001). E57, o63–o65 [doi:10.1107/S1600536800019097]
A 2-styrylboronate ester
William Clegg, Todd B. Marder, Andrew J. Scott, Christian Wiesauer and Walter Weissensteiner
S1. Comment
Alkenylboronic esters and acids are versatile synthetic intermediates (Lane & Kabalka, 1976) for products such as cis
-alkenes and halo-alkenes, and they have important applications in palladium-catalysed cross-coupling reactions and the
formation of organometallic compounds (Suzuki, 1985, 1998; Ohe et al., 1988; Larock et al., 1972; Pappo & Collins,
1972). The title compound, (I), was obtained by a hydroboration reaction of phenylacetylene with catecholborane (Brown
& Gupta, 1972, 1975; Brown & Chandrasekharan, 1983). The first reported preparation of this compound (Joy et al.,
1966) involved treatment of cyclooctatetraene with boron trichloride, followed by reaction of the resulting
(E)-styrylboron dichloride with catechol. In order to study the influence of electron-donating and electron-withdrawing
substituents at the para-position of the phenyl ring on the reactivity of the double bond in catalysed hydroborations of
styrylboronate esters, a series of para-substituted analogues has been prepared (Wiesauer, 1997).
The molecule (Fig. 1) is essentially completely planar, with an r.m.s. deviation of 0.071 Å from the least-squares plane
through all atoms. All torsion angles are close to 0 and 180°, the largest corresponding to a twist of about 6° about the C2
—C21 bond linking the phenyl group to the central alkene unit. Bond lengths and angles are normal, with B—C
somewhat shorter and C═C longer than generally observed for single and double bonds, respectively (Allen et al., 1992);
together with the planarity of the molecule, they indicate extensive delocalization.
It is instructive to compare the title compound with two closely related styryl bis(boronate) esters, (Z)-[(4-NC—C6H4
)-C(Bcat)═CH(Bcat)] [(II); Bcat is 1,3,2-benzodioxaborole; cat = 1,2-O2C6H4; Clegg et al., 1996] and (Z)-[(4-MeO-C6H4
)-C(Bcat)═CH(BCat)] [(III); Lesley et al., 1996]. These differ from (I) in having either a π-acceptor or π-donor substituent
at the para-position of the phenyl ring attached to the α-carbon of the alkene and also a second Bcat moiety at Cα. In (II)
and (III), the Bcat at the β-position is approximately coplanar (dihedral angles approximately 9 and 7°, respectively) with
the alkene unit, as in (I), whereas the Bcat groups at Cα in (II) and (III) are roughly perpendicular (80 and 84°,
respectively) to the alkene plane. In both (II) and (III), the Cβ—B distances [1.535 (2) and 1.526 (2) Å] are significantly
shorter than their Cα—B distances [1.569 (2) and 1.566 (2) Å] and are consistent with the Cβ—B distance in (I) of
1.5321 (17) Å. Presumably this is a reflection of the delocalization present when the Bcat group is coplanar with the
alkene and trans to the aryl moiety. The C═C distance in (I) [1.3368 (16) Å] is shorter than the corresponding distance in
either (II) or (III) [1.349 (2) and 1.348 (2) Å, respectively] and is probably a result of less steric repulsion in the less
substituted alkene.
This structure was recently used as a test of crystal structure prediction methods (Lommerse et al., 2000), and proved to
be a considerable challenge to the currently available software systems, with only one prediction reasonably close to the
supporting information
sup-2
Acta Cryst. (2001). E57, o63–o65 S2. Experimental
In a nitrogen-filled glove-box, phenylacetylene (4.0 g, 39 mmol) and catecholborane (4.7 g, 39 mmol) were heated at 343
K for 2 h in a scintillation vial. On cooling, an orange solid was formed. The pure product was obtained as a colourless
crystalline material by recrystallization from diethylether/hexane by solvent diffusion at 238 K, in 70% yield. Analysis
calculated: C 75.73, H 4.99%; found: C 75.30, H 4.98%. 1H NMR (200 MHz): δ 6.48 (d, J = 18.5 Hz, 1H, PhCH), 7.08
(m, 2H, C6H4), 7.26 (m, 2H, C6H4), 7.38 (m, 3H, C6H5), 7.58 (m, 2H, C6H5), 7.77 (d, J = 18.5 Hz, 1H, BCH). 13C{1H}
NMR (50 MHz): δ 112.3 (2 C, C3 and C6 of C6H4), 122.6 (2 C, C4 and C5 of C6H4), 127.4 (2 C, Ph), 128.7 (2 C, Ph), 129.6
(1 C, C4 of Ph), 136.9 (1 C, C1 of Ph), 148.3 (2 C, C1 and C2 of C6H4), 152.0 (1 C, PhCH), resonances of C bonded to B
too broad to be observed. 11B{1H} NMR (64 MHz): δ 31.3. MS: 222 (M+, 100%), 207 (26%), 196 (52%), 179 (30%), 178
(30%), 144 (68%), 136 (78%), 129 (21%), 120 (84%), 111 (60%), 110 (79%), 102 (77%), 92 (48%).
S3. Refinement
The data collection nominally covered over a hemisphere of reciprocal space, by a combination of three sets of
exposures; each set had a different φ angle for the crystal and each exposure covered 0.3° in ω. H atoms were placed
geometrically and refined with a riding model (including free rotation about C—C bonds), and with Uiso constrained to be
[image:5.610.111.490.322.453.2]1.2Ueq of the carrier atom.
Figure 1
The molecular structure of (I) with atom labels and 50% probability ellipsoids for non-H atoms.
(E)-2-(2-phenylethenyl)-1,3,2-benzodioxaborole
Crystal data
C14H11BO2
Mr = 222.04
Monoclinic, P21/c
a = 6.8351 (9) Å
b = 7.6342 (10) Å
c = 21.422 (3) Å
β = 96.447 (3)°
V = 1110.8 (3) Å3
Z = 4
F(000) = 464
Dx = 1.328 Mg m−3
Mo Kα radiation, λ = 0.71073 Å Cell parameters from 5221 reflections
θ = 2.8–28.6°
µ = 0.09 mm−1
T = 160 K Block, colourless 0.62 × 0.50 × 0.40 mm
Data collection
Bruker SMART 1K CCD diffractometer
Radiation source: sealed tube Graphite monochromator
Detector resolution: 8.192 pixels mm-1
ω rotation with narrow frames scans 6709 measured reflections
supporting information
sup-3
Acta Cryst. (2001). E57, o63–o65
2326 reflections with I > 2σ(I)
Rint = 0.022
θmax = 28.6°, θmin = 1.9°
h = −9→8
k = −6→10
l = −28→28
Refinement
Refinement on F2 Least-squares matrix: full
R[F2 > 2σ(F2)] = 0.037
wR(F2) = 0.095
S = 1.06 2571 reflections 155 parameters 0 restraints
Primary atom site location: structure-invariant direct methods
Secondary atom site location: difference Fourier map
Hydrogen site location: inferred from neighbouring sites
H-atom parameters constrained
w = 1/[σ2(F
o2) + (0.0389P)2 + 0.3966P] where P = (Fo2 + 2Fc2)/3
(Δ/σ)max = 0.001 Δρmax = 0.26 e Å−3 Δρmin = −0.15 e Å−3
Extinction correction: SHELXTL, Fc*=kFc[1+0.001xFc2λ3/sin(2θ)]-1/4 Extinction coefficient: 0.021 (2)
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
x y z Uiso*/Ueq
C1 0.28827 (17) 0.60351 (16) 0.42739 (5) 0.0297 (3)
H1 0.1941 0.5390 0.4006 0.036*
C2 0.47581 (16) 0.59715 (14) 0.41531 (5) 0.0259 (2)
H2 0.5685 0.6606 0.4430 0.031*
B1 0.21424 (19) 0.70732 (17) 0.48127 (6) 0.0273 (3)
O1 0.33111 (11) 0.80614 (10) 0.52550 (4) 0.0274 (2)
O2 0.01719 (12) 0.71233 (11) 0.49180 (4) 0.0284 (2)
C11 0.20129 (15) 0.87370 (14) 0.56460 (5) 0.0242 (2)
C12 0.01226 (16) 0.81736 (14) 0.54409 (5) 0.0244 (2)
C13 −0.14856 (16) 0.86350 (15) 0.57367 (5) 0.0284 (2)
H13 −0.2778 0.8253 0.5590 0.034*
C14 −0.11031 (18) 0.96985 (15) 0.62656 (5) 0.0303 (3)
H14 −0.2163 1.0045 0.6489 0.036*
C15 0.07932 (18) 1.02632 (15) 0.64730 (5) 0.0308 (3)
H15 0.0998 1.0983 0.6836 0.037*
C16 0.24022 (17) 0.98000 (15) 0.61615 (5) 0.0290 (2)
H16 0.3696 1.0198 0.6299 0.035*
C21 0.55373 (15) 0.50245 (14) 0.36375 (5) 0.0232 (2)
C22 0.75132 (16) 0.52442 (15) 0.35424 (5) 0.0273 (2)
H22 0.8336 0.5975 0.3818 0.033*
C23 0.82871 (17) 0.44080 (16) 0.30499 (5) 0.0310 (3)
H23 0.9625 0.4589 0.2986 0.037*
C24 0.71196 (18) 0.33125 (16) 0.26518 (5) 0.0303 (3)
H24 0.7654 0.2734 0.2317 0.036*
C25 0.51591 (17) 0.30617 (15) 0.27446 (5) 0.0295 (3)
H25 0.4354 0.2306 0.2473 0.035*
C26 0.43755 (16) 0.39075 (15) 0.32302 (5) 0.0266 (2)
supporting information
sup-4
Acta Cryst. (2001). E57, o63–o65 Atomic displacement parameters (Å2)
U11 U22 U33 U12 U13 U23
C1 0.0290 (6) 0.0314 (6) 0.0286 (6) 0.0024 (4) 0.0026 (4) −0.0029 (5)
C2 0.0299 (5) 0.0247 (5) 0.0229 (5) 0.0012 (4) 0.0017 (4) 0.0011 (4)
B1 0.0278 (6) 0.0272 (6) 0.0270 (6) 0.0034 (5) 0.0034 (5) 0.0025 (5)
O1 0.0242 (4) 0.0310 (4) 0.0275 (4) 0.0029 (3) 0.0045 (3) 0.0003 (3)
O2 0.0267 (4) 0.0309 (4) 0.0279 (4) 0.0006 (3) 0.0042 (3) −0.0055 (3)
C11 0.0245 (5) 0.0236 (5) 0.0249 (5) 0.0032 (4) 0.0047 (4) 0.0047 (4)
C12 0.0289 (5) 0.0217 (5) 0.0226 (5) 0.0013 (4) 0.0027 (4) 0.0012 (4)
C13 0.0251 (5) 0.0293 (6) 0.0312 (6) 0.0006 (4) 0.0051 (4) 0.0012 (5)
C14 0.0334 (6) 0.0283 (6) 0.0309 (6) 0.0042 (5) 0.0105 (5) 0.0012 (5)
C15 0.0396 (6) 0.0273 (5) 0.0256 (5) 0.0003 (5) 0.0045 (4) −0.0016 (4)
C16 0.0298 (6) 0.0282 (6) 0.0283 (5) −0.0018 (4) 0.0000 (4) 0.0013 (4)
C21 0.0244 (5) 0.0230 (5) 0.0220 (5) 0.0029 (4) 0.0023 (4) 0.0040 (4)
C22 0.0248 (5) 0.0288 (5) 0.0276 (5) −0.0003 (4) 0.0006 (4) 0.0018 (4)
C23 0.0248 (5) 0.0362 (6) 0.0330 (6) 0.0026 (5) 0.0077 (4) 0.0044 (5)
C24 0.0368 (6) 0.0293 (6) 0.0259 (5) 0.0059 (5) 0.0087 (4) 0.0016 (4)
C25 0.0338 (6) 0.0273 (6) 0.0270 (5) −0.0003 (4) 0.0010 (4) −0.0017 (4)
C26 0.0240 (5) 0.0275 (5) 0.0284 (5) −0.0009 (4) 0.0031 (4) 0.0002 (4)
Geometric parameters (Å, º)
C1—C2 1.3368 (16) C14—H14 0.950
C1—B1 1.5321 (17) C15—C16 1.3945 (17)
C1—H1 0.950 C15—H15 0.950
C2—C21 1.4692 (15) C16—H16 0.950
C2—H2 0.950 C21—C22 1.3983 (15)
B1—O2 1.3909 (15) C21—C26 1.4013 (15)
B1—O1 1.3910 (15) C22—C23 1.3877 (16)
O1—C11 1.3864 (13) C22—H22 0.950
O2—C12 1.3811 (13) C23—C24 1.3823 (17)
C11—C16 1.3722 (16) C23—H23 0.950
C11—C12 1.3856 (15) C24—C25 1.3898 (16)
C12—C13 1.3749 (15) C24—H24 0.950
C13—C14 1.3944 (16) C25—C26 1.3824 (16)
C13—H13 0.950 C25—H25 0.950
C14—C15 1.3907 (17) C26—H26 0.950
C2—C1—B1 124.72 (11) C14—C15—H15 119.2
C2—C1—H1 117.6 C16—C15—H15 119.2
B1—C1—H1 117.6 C11—C16—C15 116.29 (11)
C1—C2—C21 126.86 (11) C11—C16—H16 121.9
C1—C2—H2 116.6 C15—C16—H16 121.9
C21—C2—H2 116.6 C22—C21—C26 118.12 (10)
O2—B1—O1 111.54 (10) C22—C21—C2 119.22 (10)
O2—B1—C1 122.96 (11) C26—C21—C2 122.66 (10)
supporting information
sup-5
Acta Cryst. (2001). E57, o63–o65
C11—O1—B1 104.82 (9) C23—C22—H22 119.6
C12—O2—B1 105.06 (9) C21—C22—H22 119.6
C16—C11—C12 121.90 (10) C24—C23—C22 120.29 (10)
C16—C11—O1 128.82 (10) C24—C23—H23 119.9
C12—C11—O1 109.28 (9) C22—C23—H23 119.9
C13—C12—O2 128.04 (10) C23—C24—C25 119.63 (10)
C13—C12—C11 122.66 (10) C23—C24—H24 120.2
O2—C12—C11 109.30 (9) C25—C24—H24 120.2
C12—C13—C14 115.91 (11) C26—C25—C24 120.29 (11)
C12—C13—H13 122.0 C26—C25—H25 119.9
C14—C13—H13 122.0 C24—C25—H25 119.9
C15—C14—C13 121.54 (11) C25—C26—C21 120.83 (10)
C15—C14—H14 119.2 C25—C26—H26 119.6
C13—C14—H14 119.2 C21—C26—H26 119.6
C14—C15—C16 121.69 (11)
B1—C1—C2—C21 −178.83 (11) C11—C12—C13—C14 −0.71 (17)
C2—C1—B1—O2 179.17 (11) C12—C13—C14—C15 0.58 (17)
C2—C1—B1—O1 −1.22 (19) C13—C14—C15—C16 0.25 (18)
O2—B1—O1—C11 0.16 (12) C12—C11—C16—C15 0.84 (17)
C1—B1—O1—C11 −179.49 (11) O1—C11—C16—C15 −179.21 (10)
O1—B1—O2—C12 0.00 (12) C14—C15—C16—C11 −0.96 (17)
C1—B1—O2—C12 179.65 (10) C1—C2—C21—C22 173.52 (11)
B1—O1—C11—C16 179.80 (11) C1—C2—C21—C26 −6.08 (18)
B1—O1—C11—C12 −0.25 (12) C26—C21—C22—C23 1.36 (16)
B1—O2—C12—C13 −179.91 (11) C2—C21—C22—C23 −178.25 (10)
B1—O2—C12—C11 −0.16 (12) C21—C22—C23—C24 −1.30 (17)
C16—C11—C12—C13 −0.01 (17) C22—C23—C24—C25 0.46 (18)
O1—C11—C12—C13 −179.97 (10) C23—C24—C25—C26 0.28 (17)
C16—C11—C12—O2 −179.78 (10) C24—C25—C26—C21 −0.20 (17)
O1—C11—C12—O2 0.26 (12) C22—C21—C26—C25 −0.62 (16)