organic papers
Acta Cryst.(2006). E62, o2365–o2366 doi:10.1107/S1600536806017211 De-Suo Yang C
13H11BrN2O
o2365
Acta Crystallographica Section E Structure Reports Online
ISSN 1600-5368
4-Bromo-2-[(5-methylpyridin-2-ylimino)-methyl]phenol
De-Suo Yang
Department of Chemistry and Chemical Engineering, Baoji College of Arts and Sciences, Baoji 721007, People’s Republic of China
Correspondence e-mail: desuoyang@yahoo.com.cn
Key indicators
Single-crystal X-ray study T= 298 K
Mean(C–C) = 0.008 A˚ Rfactor = 0.044 wRfactor = 0.117
Data-to-parameter ratio = 15.7
For details of how these key indicators were automatically derived from the article, see http://journals.iucr.org/e.
Received 8 May 2006 Accepted 9 May 2006
#2006 International Union of Crystallography All rights reserved
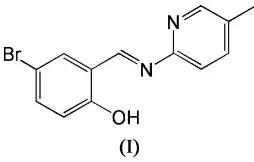
The molecule of the title compound, C13H11BrN2O, displays a
transconfiguration with respect to the central C N double bond and is almost planar. An intramolecular O—H N hydrogen bond [O N = 2.589 (6) A˚ ] may influence the overall conformation.
Comment
Schiff compounds play an important role in the development of coordination chemistry (Telferet al., 2004; Ramnauthet al., 2004; Musieet al., 2001; Bernardoet al., 1996; Paulet al., 2002). Two crystal structures of Schiff base compounds have recently been reported by the author (Yang, 2006a,b). As an extension of work on the structural characterization of such compounds, the crystal structure of the title compound, (I), is reported here (Fig. 1).
In (I), all the bond lengths are within normal ranges (Allen
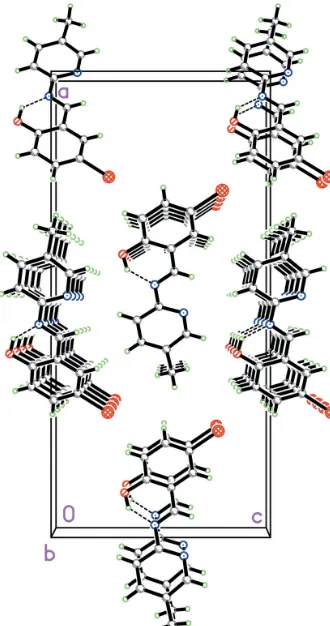
[image:1.610.267.394.369.450.2] [image:1.610.208.459.591.707.2]et al., 1987) and comparable to the corresponding values observed in the similar crystal structures reported recently (Yang, 2006a), e.g.the central transC7 N1 bond length of 1.269 (7) A˚ . The molecule is nearly planar, with a dihedral angle of 4.0 (5) between the benzene ring and the pyridine ring. An intramolecular O—H N hydrogen bond (Table 1) may influence the overall conformation. In the crystal struc-ture, the molecular packing in (I) is stabilized only by van der
Figure 1
Waals interactions (Fig. 2) as there are no intermolecular hydrogen bonds or significant–stacking interactions.
Experimental
Reagents and solvents used were of commercially available quality. 5-Bromosalicylaldehyde (0.1 mmol, 20.1 mg) and 5-methyl-2-amino-pyridine (0.1 mmol, 10.8 mg) were dissolved in MeOH (10 ml). The mixture was stirred at 298 K to give a clear yellow solution. Crystals of (I) were formed by slow evaporation of the solvent over a period of about 10 d at 298 K.
Crystal data
C13H11BrN2O
Mr= 291.15 Orthorhombic,Pna21
a= 24.245 (4) A˚
b= 4.358 (1) A˚
c= 11.401 (2) A˚
V= 1204.6 (4) A˚3
Z= 4
Dx= 1.605 Mg m 3 MoKradiation
= 3.40 mm 1
T= 298 (2) K Block, yellow 0.200.180.18 mm
Data collection
Bruker APEX area-detector diffractometer
’and!scans
Absorption correction: multi-scan (SADABS; Bruker, 2002)
Tmin= 0.517,Tmax= 0.545
8591 measured reflections 2456 independent reflections 1618 reflections withI> 2(I)
Rint= 0.056
max= 26.5
Refinement
Refinement onF2
R[F2> 2(F2)] = 0.044
wR(F2) = 0.117
S= 1.07 2456 reflections 156 parameters
H-atom parameters constrained
w= 1/[2(F
o2) + 0.5825P] whereP= (Fo2+ 2Fc2)/3 (/)max< 0.001
max= 0.33 e A˚ 3 min= 0.33 e A˚ 3
Absolute structure: Flack (1983), 1141 Friedel pairs.
Flack parameter: 0.02 (2)
Table 1
Hydrogen-bond geometry (A˚ ,).
D—H A D—H H A D A D—H A
O1—H1 N1 0.82 1.93 2.589 (6) 137
All H atoms were placed in idealized positions and constrained to ride on their parent atoms. Constrained distances: O—H = 0.82 A˚ , and C—H = 0.93 and 0.96 A˚ for methyl and aromatic CH groups, respectively. Isotropic displacement parameters were fixed atUiso(H)
= 1.2Uiso(C) for aromatic CH groups and 1.5Uiso(C,O) for other H
atoms.
Data collection:SMART(Bruker, 2002); cell refinement:SAINT
(Bruker, 2002); data reduction: SAINT; program(s) used to solve structure:SHELXS97(Sheldrick, 1997a); program(s) used to refine structure: SHELXL97 (Sheldrick, 1997a); molecular graphics:
SHELXTL(Sheldrick, 1997b); software used to prepare material for publication:SHELXL97.
Financial support from the Baoji College of Arts and Sciences research funds is gratefully acknowledged.
References
Allen, F. H., Kennard, O., Watson, D. G., Brammer, L., Orpen, A. G. & Taylor, R. (1987).J. Chem. Soc. Perkin Trans. 2, pp. S1–19.
Bernardo, K., Leppard, S., Robert, A., Commenges, G., Dahan, F. & Meunier, B. (1996).Inorg. Chem.35, 387–396.
Bruker (2002).SAINT(Version 6.36A,SMART(Version 5.10) andSADABS
(Version 2.01). Bruker AXS Inc., Madison, Wisconsin, USA. Flack, H. D. (1983).Acta Cryst.A39, 876–881.
Musie, G. T., Wei, M., Subramaniam, B. & Busch, D. H. (2001).Inorg. Chem.
40, 3336–3341.
Paul, S., Barik, A. K., Peng, S. M. & Kar, S. K. (2002).Inorg. Chem.41, 5803– 5809.
Ramnauth, R., Al-Juaid, S., Motevalli, M., Parkin, B. C. & Sullivan, A. C. (2004).Inorg. Chem.43, 4072–4079.
Sheldrick, G. M. (1997a). SHELXL97 and SHELXS97. University of Go¨ttingen, Germany.
Sheldrick, G. M. (1997b).SHELXTL. Version 5.1. Bruker AXS Inc., Madison, Wisconsin, USA.
Telfer, S. G., Sato, T., Harada, T., Kuroda, R., Lefebvre, J. & Leznoff, D. B. (2004).Inorg. Chem.43, 6168–6176.
[image:2.610.357.522.72.385.2]Yang, D.-S. (2006a).Acta Cryst.E62, o1395–o1396. Yang, D.-S. (2006b).Acta Cryst.E62, o1591–o1592.
Figure 2
supporting information
sup-1 Acta Cryst. (2006). E62, o2365–o2366
supporting information
Acta Cryst. (2006). E62, o2365–o2366 [https://doi.org/10.1107/S1600536806017211]
4-Bromo-2-[(5-methylpyridin-2-ylimino)methyl]phenol
De-Suo Yang
4-Bromo-2-[(5-methylpyridin-2-ylimino)methyl]phenol
Crystal data
C13H11BrN2O Mr = 291.15
Orthorhombic, Pna21 Hall symbol: P 2c -2n a = 24.245 (4) Å b = 4.358 (1) Å c = 11.401 (2) Å V = 1204.6 (4) Å3 Z = 4
F(000) = 584 Dx = 1.605 Mg m−3
Mo Kα radiation, λ = 0.71073 Å Cell parameters from 1681 reflections θ = 2.4–24.5°
µ = 3.40 mm−1 T = 298 K Block, yellow
0.20 × 0.18 × 0.18 mm
Data collection
Bruker APEX area-detector diffractometer
Radiation source: fine-focus sealed tube Graphite monochromator
φ and ω scans
Absorption correction: multi-scan (SADABS; Bruker, 2002) Tmin = 0.517, Tmax = 0.545
8591 measured reflections 2456 independent reflections 1618 reflections with I > 2σ(I) Rint = 0.056
θmax = 26.5°, θmin = 1.7° h = −29→29
k = −5→5 l = −14→13
Refinement
Refinement on F2 Least-squares matrix: full R[F2 > 2σ(F2)] = 0.044 wR(F2) = 0.117 S = 1.07 2456 reflections 156 parameters 1 restraint
Primary atom site location: structure-invariant direct methods
Secondary atom site location: difference Fourier map
Hydrogen site location: inferred from neighbouring sites
H-atom parameters constrained w = 1/[σ2(F
o2) + 0.5825P] where P = (Fo2 + 2Fc2)/3 (Δ/σ)max < 0.001
Δρmax = 0.33 e Å−3 Δρmin = −0.33 e Å−3
Absolute structure: Flack (1983), 1141 Friedel pairs.
Absolute structure parameter: 0.02 (2)
Special details
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger.
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
x y z Uiso*/Ueq
Br1 0.72749 (2) 1.58944 (15) 0.76403 (9) 0.0700 (3)
O1 0.60188 (19) 1.1807 (10) 0.3385 (3) 0.0593 (12)
H1 0.5712 1.1158 0.3562 0.089*
N1 0.5406 (2) 0.8508 (10) 0.4752 (4) 0.0416 (12)
N2 0.4823 (2) 0.5713 (12) 0.6027 (4) 0.0495 (12)
C1 0.6135 (2) 1.1646 (11) 0.5476 (5) 0.0388 (13)
C2 0.6286 (2) 1.2696 (13) 0.4362 (5) 0.0450 (14)
C3 0.6724 (2) 1.4721 (13) 0.4246 (6) 0.0483 (15)
H3 0.6820 1.5460 0.3508 0.058*
C4 0.7018 (3) 1.5641 (13) 0.5220 (6) 0.0505 (15)
H4 0.7321 1.6936 0.5138 0.061*
C5 0.6863 (3) 1.4644 (13) 0.6300 (5) 0.0462 (15)
C6 0.6426 (3) 1.2626 (13) 0.6449 (5) 0.0441 (14)
H6 0.6330 1.1943 0.7194 0.053*
C7 0.5675 (2) 0.9587 (12) 0.5614 (5) 0.0420 (14)
H7 0.5570 0.9022 0.6368 0.050*
C8 0.4956 (2) 0.6523 (13) 0.4931 (5) 0.0414 (14)
C9 0.4673 (3) 0.5490 (15) 0.3959 (5) 0.0510 (16)
H9 0.4787 0.6008 0.3206 0.061*
C10 0.4209 (3) 0.3645 (14) 0.4137 (5) 0.0516 (16)
H10 0.4007 0.2950 0.3496 0.062*
C11 0.4050 (2) 0.2849 (13) 0.5259 (5) 0.0463 (14)
C12 0.4381 (3) 0.3945 (14) 0.6161 (5) 0.0507 (16)
H12 0.4284 0.3398 0.6922 0.061*
C13 0.3556 (3) 0.0966 (16) 0.5508 (7) 0.069 (2)
H13A 0.3244 0.2285 0.5628 0.104*
H13B 0.3618 −0.0239 0.6200 0.104*
H13C 0.3485 −0.0371 0.4856 0.104*
Atomic displacement parameters (Å2)
U11 U22 U33 U12 U13 U23
Br1 0.0634 (4) 0.0781 (4) 0.0684 (4) −0.0050 (3) −0.0172 (5) −0.0138 (5)
O1 0.073 (3) 0.069 (3) 0.036 (2) −0.008 (2) 0.000 (2) −0.001 (2)
N1 0.049 (3) 0.037 (3) 0.038 (3) −0.002 (2) 0.002 (2) 0.001 (2)
N2 0.057 (3) 0.059 (3) 0.033 (3) −0.006 (3) 0.004 (2) 0.000 (2)
C1 0.049 (3) 0.033 (3) 0.035 (3) 0.006 (3) 0.006 (3) −0.001 (2)
C2 0.056 (4) 0.046 (3) 0.033 (3) 0.012 (3) 0.002 (3) −0.006 (3)
C3 0.048 (4) 0.043 (3) 0.053 (4) 0.006 (3) 0.013 (3) 0.003 (3)
supporting information
sup-3 Acta Cryst. (2006). E62, o2365–o2366
C5 0.047 (4) 0.037 (3) 0.055 (4) 0.008 (3) −0.004 (3) −0.012 (3)
C6 0.052 (4) 0.042 (3) 0.039 (3) 0.004 (3) −0.005 (3) 0.002 (3)
C7 0.053 (4) 0.040 (3) 0.033 (3) 0.002 (3) 0.001 (3) 0.007 (3)
C8 0.049 (4) 0.045 (3) 0.031 (3) 0.008 (3) 0.002 (3) −0.004 (2)
C9 0.053 (4) 0.066 (4) 0.034 (3) −0.006 (3) 0.003 (3) 0.002 (3)
C10 0.057 (4) 0.057 (4) 0.041 (3) 0.006 (3) 0.000 (3) −0.003 (3)
C11 0.050 (4) 0.044 (3) 0.045 (4) 0.004 (3) −0.002 (3) −0.001 (3)
C12 0.061 (4) 0.059 (4) 0.032 (3) −0.004 (3) 0.001 (3) 0.004 (3)
C13 0.052 (4) 0.082 (5) 0.073 (5) −0.018 (4) −0.005 (4) 0.020 (4)
Geometric parameters (Å, º)
Br1—C5 1.905 (6) C5—C6 1.388 (9)
O1—C2 1.346 (7) C6—H6 0.9300
O1—H1 0.8200 C7—H7 0.9300
N1—C7 1.269 (7) C8—C9 1.378 (8)
N1—C8 1.408 (7) C9—C10 1.396 (9)
N2—C12 1.330 (7) C9—H9 0.9300
N2—C8 1.338 (7) C10—C11 1.381 (8)
C1—C6 1.381 (8) C10—H10 0.9300
C1—C2 1.399 (8) C11—C12 1.388 (8)
C1—C7 1.440 (8) C11—C13 1.480 (8)
C2—C3 1.387 (8) C12—H12 0.9300
C3—C4 1.380 (9) C13—H13A 0.9600
C3—H3 0.9300 C13—H13B 0.9600
C4—C5 1.359 (9) C13—H13C 0.9600
C4—H4 0.9300
C2—O1—H1 109.5 C1—C7—H7 118.5
C7—N1—C8 120.9 (5) N2—C8—C9 123.1 (5)
C12—N2—C8 117.0 (5) N2—C8—N1 118.9 (5)
C6—C1—C2 119.6 (5) C9—C8—N1 118.1 (5)
C6—C1—C7 120.0 (5) C8—C9—C10 118.2 (5)
C2—C1—C7 120.4 (5) C8—C9—H9 120.9
O1—C2—C3 118.2 (5) C10—C9—H9 120.9
O1—C2—C1 122.1 (5) C11—C10—C9 120.3 (6)
C3—C2—C1 119.7 (5) C11—C10—H10 119.9
C4—C3—C2 120.2 (6) C9—C10—H10 119.9
C4—C3—H3 119.9 C10—C11—C12 116.0 (6)
C2—C3—H3 119.9 C10—C11—C13 122.9 (6)
C5—C4—C3 119.6 (6) C12—C11—C13 121.1 (6)
C5—C4—H4 120.2 N2—C12—C11 125.5 (5)
C3—C4—H4 120.2 N2—C12—H12 117.3
C4—C5—C6 121.6 (6) C11—C12—H12 117.3
C4—C5—Br1 119.4 (5) C11—C13—H13A 109.5
C6—C5—Br1 118.9 (4) C11—C13—H13B 109.5
C1—C6—C5 119.2 (5) H13A—C13—H13B 109.5
C5—C6—H6 120.4 H13A—C13—H13C 109.5
N1—C7—C1 123.0 (5) H13B—C13—H13C 109.5
N1—C7—H7 118.5
Hydrogen-bond geometry (Å, º)
D—H···A D—H H···A D···A D—H···A