1Department of Orthopaedic Surgery, University of California, San Francisco, CA, USA. 2Materials Sciences Division, Lawrence Berkeley National Laboratory, Berkeley, CA, USA. 3Department of Mechanical Engineering, University of Utah, Salt Lake City, UT, USA. 4AO Research Institute Davos, Davos-Platz, Switzerland. 5SCANCO Medical AG, Bruettisellen, Switzerland. 6Laboratory of Biomechanical Orthopedics, Institute of Bioengineering, Swiss Federal Institute of Technology Lausanne, EPFL, Lausanne, Switzerland. 7Department of Materials Science and Engineering, University of California, Berkeley, CA, USA. Tamara Alliston and Robert O. Ritchie contributed equally to this work. *e-mail: [email protected]
F
or engineered materials (such as metals, ceramics and compos-ites), fatigue is by far the most common cause of failure. In frac-ture-mechanics terms, fatigue implies the decrease of a material’s mechanical resistance as a result of the repetitive (cyclic) application of a load that by itself is not sufficient to cause the static failure of the material. Fatigue failure is defined as the number of cycles or time needed to reach a predefined level of material damage or crack growth. In the case of an engineered structure (such as a bridge), the structure is not only designed to resist a maximum static load but, more importantly, to also support a certain number of loading cycles (caused by daily traffic, for example) before it requires repair. Although these concepts are widely and practically implemented by civil engineers, the same ideas have not yet been thoroughly inte-grated into the understanding of bone as a structural material, or into the clinical prevention of fragility fractures in human populations. In bone research, the predominant fracture mechanism is still uncer-tain: are bones more prone to fracture by a fatigue mechanism under cyclic loading, as are most engineered materials, or are they more inclined to break in a static-fracture mode by a single overload, as has been implied in a majority of the research on bone fragility1?Persuasive evidence that the mechanisms of fatigue-driven crack growth are extensively involved in bone fracture began to emerge approximately a century ago. At that time, the medical term ‘stress fracture’ (Box 1) was applied to describe injuries associated with military personnel and athletes who abruptly engaged in a regimen of marching or running2–4. Such injuries are now clinically termed fatigue fractures (Box 1), and are now recognized as one of the most common type of fractures involved in all sports injuries (~0.7 to 20%)5,6. Although most research on fatigue-induced fracture was dedicated to study healthy bone from soldiers and athletes7–11, another type of fatigue-induced fracture, clinically termed insufficiency frac-tures (Box 1), is now becoming more apparent. Insufficiency frac-tures are caused by habitual daily loading in the elderly (which, in the context of this Perspective, we define as people older than approxi-mately 50 years) or in patients with bone fragility12–16.
In healthy bone, microdamage accumulates slowly during cyclic loading and has sufficient time to undergo repair17–20. In contrast, either the rapid growth of fatigue microdamage or insufficient repair eventually causes catastrophic failure18,20. In age-related and bone-fragility diseases, factors such as changes in bone mass, in bone quality at multiple length scales and in bone repair rates pro-vide the basis for increased accumulation of damage that leads to insufficiency fractures21–23.
Improved imaging techniques have increased clinical aware-ness in that a proportion of fragility fractures might actually be insufficiency fractures24. Insufficiency fractures are most often associated with osteoporosis, but are also apparent in diseases such as rheumatoid arthritis, Paget’s disease, osteomalacia, osteo-genesis imperfecta and diabetes25. Certain therapies, such as pelvic irradiation and drug treatment, or poor nutrition and hormonal imbalance, can also promote insufficiency fractures. Even rare but serious complications of long-term bisphosphonate treatment (the controversial ‘atypical femoral fractures’) are thought to be insufficiency fractures26–30. However, quantifying the extent to which fractures of cortical bone result from cyclic fatigue dam-age in live individuals is a difficult exercise. Fatigue mechanisms occur at nanometre to micrometre length scales over long peri-ods of time, and involve numerous confounding factors, such as loading history and patient history, bone mass, bone quality and cellular bone repair.
The diversity of fractures observed in human bone is not cur-rently matched by an equally sophisticated understanding of the underlying biological or mechanical mechanisms. Improved under-standing could impact the clinical approach to diagnosis or treat-ment. Therefore, in an attempt to explore the role of fatigue in bone, in this Perspective we raise and discuss the following points:
t To what extent does daily cyclic loading (rather than single overload events) contribute to fragility fractures? How can such fatigue damage be detected and characterized?
Fatigue as the missing link between bone fragility
and fracture
Claire Acevedo
1,2,3*, Vincent A. Stadelmann
4,5, Dominique P. Pioletti
6, Tamara Alliston
1and
Robert O. Ritchie
2,7The prevention of fragility fractures in bone—pathologic fractures resulting from daily activity and mostly occurring in the elderly population—has been a long-term clinical quest. Recent research indicating that falls in the elderly might be the conse-quence of fracture rather than its cause has raised fundamental questions about the origin of fragility fractures. Is day-to-day cyclic loading, instead of a single-load event such as a fall, the main cause of progressively growing fractures? Are fragility fractures predominantly affected by bone quality rather than bone mass, which is the clinical indicator of fracture risk? Do osteocytes actively participate in the bone repair process? In this Perspective, we discuss the central role of cyclic fatigue in bone fragility fracture.
t To what extent does the reduced quality of fragile bone affect fatigue resistance?
t What are the cellular mechanisms by which fatigue damage stimulates local repair? What is the role of osteocytes in such repair? How do these repair processes vary with time?
t Does microdamage resulting from cyclic loading prevent or promote stress fractures?
The emerging awareness of insufficiency fractures
The early stages of an insufficiency fracture are accompanied by peri-osteal reaction oedema and bone marrow oedema, followed by the appearance of subtle linear fracture lines and the formation of a cal-lus to heal the fracture site31. Clinically, most insufficiency fractures are reported in patients over 60 years of age, especially in women with postmenopausal osteoporosis25, 32,33. However, the true preva-lence of insufficiency fractures in the elderly is unknown. The lack of a systematic assessment of their incidence is due to three main reasons. First, although insufficiency fractures have a distinct fail-ure pattern, they do not have a diagnosis code of their own. Several clinical terms are used to describe insufficiency fractures, and these terms are often interchanged. These include spontaneous fractures, minimal-trauma fractures, low-energy fractures, pathologic frac-tures, fragility fractures and compression fractures. This confusion in terminology illustrates one difficulty in identifying insufficiency fractures. Second, the primary clinical symptom of insufficiency fractures, pain, is often vague and can easily be attributed to other pathologic conditions in the elderly. Consequently, physicians do not always make the link between pain and bone fragility resulting from insufficiency fracture14. The third reason is that the early stages of stress fracture, characterized by oedema and hairline cracks resulting
from fatigue damage, are particularly difficult to detect with ordi-nary radiographic imaging. Alternative imaging techniques—such as magnetic resonance imaging (MRI), high-resolution computed tomography, bone scanning and scintigraphy25,34—are more sensitive for the detection of the associated marrow oedema but they usually do not have adequate resolution to detect a fatigue crack of a few micrometres in width35,36. Moreover, these tests are not regularly used to detect fractures in the context of advanced age. Once the critical crack size is reached and the final separation of the bone has occurred by brittle or catastrophic fracture, it is particularly challenging to dis-cern whether the fracture was the result of an insufficiency fracture induced by cyclic loading or of an overload fracture induced by a single impact. In materials science, fractographic analysis (that is, the study of fracture surfaces) would be used to determine the origin of failure; however, the technique is obviously not applicable for the visualization of bone fractures within a living organism.
The skeletal sites more commonly affected by insufficiency frac-tures are the lower limbs (tibia, fibula, calcaneus and metatarsus), spine (vertebrae) and pelvic region (sacrum, hip and coccyx)32. Interestingly, vertebrae are prone to fractures resulting from cyclic loading, whereas wrist fractures are mainly associated with single loading events37. Most insufficiency fractures in elderly patients engaged in exercise, such as marching or running, occur in the lower limbs13–15. Insufficiency fractures in the spine and pelvis are more common in bedridden individuals in nursing homes, where prolonged bed rest can induce loss of bone mass and quality as well as muscle atrophy38–40, which is known to affect the levels of strain and strain rate in older bone41,42.
Importantly, for elderly patients, insufficiency fractures are indi-cated as more likely to be the cause of a fall rather than the result of
Box 1 | Glossary of terms associated with bone fatigue and bone fracture
Cyclic loading: repeated and fluctuating loading68–70.
Fracture: complete failure of the material, caused by monotonic or cyclic loading68–70.
Fatigue: the process of initiation and propagation of cracks resulting from cyclic loading, with a stress magnitude invariably below the material’s monotonic strength68–70.
Fragility fracture (medical terminology): pathologic bone fracture that occurs when the bone is so fragile that it breaks unexpectedly as a result of daily activities. For instance, fragility fractures may happen during common events—such as falling from a standing position, coughing or sneezing—that would not have led to a broken bone in a healthy person.
Stress fracture (medical terminology): fracture of a bone caused by cyclic loading. Stress fractures comprise both fatigue fractures, which result from vigorous physical activities involving excessive cyclic loading on healthy bone, and insufficiency fractures, which result from everyday activities involving daily cyclic loading on weakened bone11, 24,125.
Microcrack: a type of microdamage characterized by a microscopic level of matrix failure in bone. Fatigue microcracks in bone are often linear20, 23,55.
Diffuse damage: a type of microdamage characterized by patches of submicroscopic cracks. Diffuse damage is usually observed at a level of fatigue damage that is lower than that leading to microcracks55.
Crack initiation (stage I): formation and accumulation of small damage associated with loss of stiffness68–70.
Stable (subcritical) crack growth (stage II): slow and incremental crack growth with each cycle of load68–70.
Unstable (critical) crack growth (stage III): crack growth causing sudden failure. This occurs when the advancing crack has reached a critical size (that is, at the critical fracture toughness Kc)68–70.
Bone mass: amount of mineralized bone material in a given volume of interest (one measure of bone quantity).
Bone quality: factors other than bone mass that contribute to the bone’s mechanical properties, such as strength and fracture resistance. Aspects of bone quality may include collagen-mineral composition, multiple-length-scale structure and bone-matrix material properties.
Bone resorption: removal of mineralized bone, traditionally associated with osteoclast activity. More recently, osteocytes have been implicated in bone resorption locally in the lacuna-canalicular space.
Bone formation: deposition of new bone matrix, usually associated with osteoblast activity and recently associated with local osteocyte activity.
Bone remodelling: the process of bone resorption followed by the formation of new bone.
it38,43–45. A prospective study of 54 elderly patients with fracture of the femoral neck concluded that perhaps a quarter of them had stress fractures, which later became fully broken43. These patients described their falls as the leg ‘giving way’ after days of growing pain. These sto-ries were directly related to another case report43 of the same charac-teristic symptoms of critical crack growth, and in which a radiograph of the hip on the day before the fall had revealed a stress fracture.
A few case series have reported the prevalence of insufficiency fractures for specific skeletal sites or in specific living conditions. For instance, the incidence of insufficiency fracture in the sacrum of the elderly has been shown to be between 1% and 5%46–49. In nurs-ing homes, between 1% and 4% of elderly patients were deemed to have suffered insufficiency fractures39,50. These numbers are likely to be underestimates, because such fractures are underdiagnosed and even undetected. Insufficiency fractures are suspected in one-third of patients with pubic fractures46,51 and in one-quarter of patients with femoral neck fractures38,43, with more than half of these patients having a history of long-bone fractures in the same or surround-ing bone38, 43,51. This suggests a delay, or even absence, of healing from a previous fracture, which leaves a local defect that then can act as the source of further fatigue cracks. Consistently, some 50% of elderly patients with pelvic insufficiency fractures had not recov-ered their former level of self-sufficiency after 45 days of hospital-ization, and 25% of them actually required institutional care40. But the most devastating aspect is that patients older than age 50 who suffer bone fractures are at a high risk of dying52,53. In fact, the risk of mortality in the elderly can increase up to eight-fold in the three months following a hip fracture (relative to age-matched and sex-matched elderly without fractures) due to complications associated with healing52. There is thus a clinical need to define the spectrum of fatigue-induced fractures more precisely to aid the prevention of these fractures, to improve the efficiency of detection and diagnosis and, more importantly, to avert the severe problems of delayed heal-ing and premature death in the elderly population.
The underrated role of cyclic loading
Most of the loads that human bodies carry daily, such as those induced by walking, running and jumping, are naturally cyclic. Distinct from these are weight-bearing loads, which are static, and impact loads, for instance those caused by falls. Human bones are naturally programmed to respond to repeated stimuli in the pro-cess of building a light and resistant skeleton54. Under physiological cyclic loading, microdamage is generated in highly stressed regions of the skeleton by the mechanism of fatigue17, 18, 23,55. Fatigue dam-age signals bone cells to repair and replace the bone matrix in these regions19,56. It has been shown that cyclic loads (but not static loads) resulting from daily activities can stimulate bone formation57,58 through this damage-repair process, even to the point that cyclic loads can accelerate fracture healing58–60.
Despite the clear importance of cyclic loading in the physiologi-cal function of healthy bone, aged or pathologic bone fracture resis-tance has primarily been studied as a single loading event, in which monotonic axial, bending or torsional testing is used to quantify bone stiffness, strength and toughness1,37. Yet insight can be gained from cyclic-loading studies of cadaveric human bones investigat-ing the relationship between age, microdamage and the number of cycles sustained before failure (defined as fatigue life). For example, age correlates with greater microdamage accumulation and with an exponential decrease in fatigue life61. In particular, older human bone forms more linear microcracks than young bone, whereas young bone tends to form more diffuse damage (that is, clusters of nanoscopic cracks) than aged bone61. This appears to be true also in in vivo loading conditions23, 62,63. Because in vivo bone has the capac-ity to undergo repair via osteocytes, osteoblasts and osteoclasts (that is, cellular repair), age-related slowdown in cellular repair com-pounds the age-dependent slowdown of the mechanical response of
bone to ex vivo cyclic loading64. This implies that, whereas in healthy bone microdamage promotes repair and prevents the formation of stress fractures, in old or diseased bone unrepaired microdamage may turn out to be the very phenomenon that drives damage accu-mulation into full fractures, as it occurs in engineered materials.
To advance the understanding of the damage mechanisms by fatigue and of the role of cyclic loading on bone fractures in healthy and fragile bones, it is necessary to explore the concepts of fatigue from the viewpoint of fracture mechanics, and compare experi-mental results from both biological and engineered materials. In engineered materials and structures, cyclic fatigue is the most ubiq-uitous mode of failure. Since it was first recognized in the nineteenth century, cyclic fatigue accounts for more than 80% of all failures, leading to catastrophic and sudden accidents such as the failure of railway axles, the collapse of metallic bridges, the failure of ships and the cracking of aircraft airframes and engines. Accordingly, devel-oping materials with enhanced resistance to fatigue, and devising strategies to estimate their safe lifetimes in the presence of fatigue loading, has become central to the design of engineered materials, and has contributed to the establishment of methods in fracture mechanics for the prevention of unexpected fractures.
The fatigue damage of engineered materials occurs in three dis-crete stages: first, damage is initiated (sometimes over short periods of time), invariably at surface imperfections or defects (stage I). Similarly, in bone fatigue, damage can initiate between the osteons in the older interstitial matrix, which is more mineralized and brittle65,66, causing a rapid drop in stiffness23. Once initiated, incipient fatigue cracks pro-gressively advance by subcritical crack growth (stage II) until a critical crack size is reached. At that point, overload fracture (or plastic col-lapse) ensues in an unstable, sometimes catastrophic, manner (stage III). This can occur when the critical stress intensity (at the critical fracture toughness Kc), is exceeded. Stages I and III can occur rapidly, the stable crack-growth stage often representing the main part of the fatigue life in engineered and biological materials. Materials that are able to tolerate the presence of damage and subsequent crack growth safely until they are repaired are regarded as ‘damage tolerant’.
In many ductile metallic and polymeric materials, the mecha-nism by which fatigue cracks progress in stage II involves successive cycles of plastic blunting during loading, followed by re-sharpening of the crack tip during unloading67. Indeed, even though the applied cyclic loads cause a nominal stress that is lower than the yield stress (that is, the elastic limit of the material), the crack-tip local stresses are invariably well above the yield stress, creating a plastic zone. These blunting or re-sharpening cycles leave characteristic mark-ings on the fracture surface of engineered materials and are known as ‘fatigue striations’68–71. Each striation is a local measure of the crack advance that occurred in that cycle, and thus a persuasive proof that the fracture occurred by cyclic fatigue.
Despite the clinical relevance of stress fractures, such a detailed mechanistic characterization of fatigue-crack growth in bone is unavailable. Nevertheless, stress fractures in bone present some common characteristics with fatigue in engineered materials. For example, transverse or short, oblique fracture lines with a smooth bone fracture surface28, 72,73 are consistent with those frequently observed in fatigued metals and alloys68–70. This can be explained by the cumulative damage model that described the cycle-dependent and time-dependent fracture modes in devitalized cortical bone74. In cycle-dependent mechanisms (fatigue mode), the repeated load-ing and unloadload-ing between maximum and minimum stresses drives crack growth. In time-dependent mechanisms (creep mode), the maximum stress itself expands the crack. The model has shown that crack-growth rates are different in the creep (time-dependent) and fatigue (cycle-dependent) modes74. Creep damage might leave a macroscopically rough crack surface following cement lines and cement bands, whereas fatigue damage is associated with smooth cracks going through the microstructural constituents, in analogy
with fatigue fracture going through the grains of the ductile met-als68–70. Cycle-dependent (and time-dependent) mechanisms of fatigue-crack advance are active in human bone75,76, and alternat-ing bluntalternat-ing and re-sharpenalternat-ing of the crack tip might be the likely mechanism of cyclic crack growth. However, the definitive charac-teristic striations associated with blunting and re-sharpening have not been reported for bone.
In bone, crack advance (stage II) can be characterized by the range of stress intensity ΔK, which characterizes the amplitude of
the local stresses and of displacements in the vicinity of a crack tip75. This can be evaluated with the well-known Paris–Erdogan law77, which relates ΔK to the crack-growth rate per cycle. The Paris–Erdogan law is extensively used for the prediction of fatigue lives in engineered materials. In addition, fatigue strength is often reported in terms of the Wöhler or S-N curve representing the stress range Δσ or S, at which failure occurs after N cycles
dur-ing experiments. These two fatigue-analysis methods—the for-mer based solely on crack growth, the latter on total life—suggest that fatigue life in bone is likely to be influenced by the loading mode65,78, the presence of initial crack-like imperfections, cyclic frequency and, most importantly, by the applied cyclic stress (or strain) range. Age-related loss of muscle strength and endurance are known to increase strain level and strain rate through reduced shock absorption41,42. Therefore, change in muscle strength will certainly cause a higher cyclic strain range and could thus play a critical role in reducing bone fatigue life. However, this is still a relatively unexplored area of bone research, as the precise role of cyclic loading and corresponding rates of bone fatigue are not well documented or understood.
The importance of collagen quality
Clinically, fracture risk is primarily assessed by bone mass or bone mineral density (BMD; Box 1) through dual-energy X-ray absorptiometry. Although bone fragility is often associated with low bone mass79, over half of all non-vertebral fractures in people over 55 years of age occurred in those with a clinically normal BMD80. Also, a ten-fold increase in fracture risk with age was found to be entirely independent of BMD81. In view of this, there is an increasing awareness that bone quality (Box 1) plays an addi-tional, if not greater, role in the occurrence of bone fragility frac-tures20,82–86. Bone quality is used to describe characteristics of the bone other than bone mass (for example, bone matrix material properties) that influence the bone’s ability to withstand applied loads and displacements. Bone quality is impaired with age, as evidenced by the increased amount of microdamage in age-ing bone61–63. Among the properties of bone, toughness is more dramatically reduced with age (a 7–12% decrease in resistance to fracture) than stiffness (1–4% decrease in resistance to elas-tic deformation) and yield strength (2–5% decrease in resistance to the onset of plastic deformation)22,87–89. These findings can be correlated with those in other studies that have shown that the accumulation of fatigue damage affects bone toughness more pro-foundly than bone stiffness and strength23. This is consistent with the finding that ageing impairs bone plasticity and the intrinsic toughening mechanisms that alter damage initiation and growth, as well as the extrinsic toughening mechanisms that dissipate deformation energy during failure90,91.
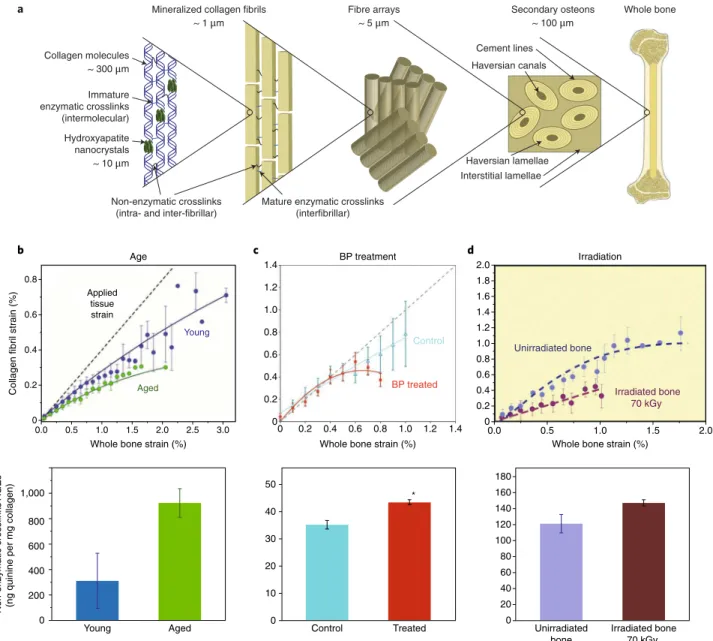
Bone derives its unique properties from its hierarchical length scales, which range from the molecular to the macroscopic, and from its graded structure (Fig. 1a). Therefore, to identify the under-lying determinants of differences in bone quality and fatigue resis-tance, it is necessary to consider the hierarchical structure of bone. At the nanoscale, the ductile collagen phase gives bone its high duc-tility and toughness, whereas the hard and brittle mineral is respon-sible for stiffness and strength. At the collagen scale, age impairs the ability of collagen fibrils to deform plastically through intrinsic
toughening mechanisms. This deficit must be compensated at higher length scales via energy dissipation through microdamage, an extrinsic toughening mechanism90. At the microscale, the osteon density is almost three times higher in aged bone than in young bone90, which gives rise to more cement lines (that is, the interface between osteon and interstitial bone tissue), where fatigue damage can form66,90. Therefore, the loss of collagen plasticity and increased osteon density contribute to the increased microdamage density with age. These findings are consistent with the reduction in crack-initiation toughness in stage I of the fatigue process. The increased osteon density also limits the potency of uncracked-ligament bridg-ing at the microscale90, which in turn degrades the ultimate crack-growth toughness during stage III.
The age-related degradation in collagen plasticity is an impor-tant factor in the loss of fatigue resistance and toughness. Fatigue-crack growth (stage II) in bone is associated with localized regions of plastic deformation in the tip of the crack, primarily taken by collagen fibril deformation. When bone material begins to deform plastically beyond its yield point, a mechanism of sliding between fibrils can ensue. Such ‘fibrillar sliding’ can maintain a constant fibril strain and, as such, enables large plastic deformations, thereby contributing to bone ductility29, 90,91. Collagen fibrils, as the constitu-ents where plastic deformations occur, are thus particularly suscep-tible to fatigue failure92,93.
Ageing and disease can cause several differences in the colla-gen environment, such as an accumulation of collacolla-gen crosslinks, variations in the bound water between the collagen and mineral phases21,94, and changes in the collagen-degrading matrix metal-loproteinases95. In addition, levels of non-collagenous proteins (osteocalcin and osteopontin)55,96 and crystal size97 contribute to differences in bone quality98. Among these factors, the increase of collagen crosslinking is a recurrent and important effect in a variety of bone fragility diseases. Accumulation of specific collagen cross-links can stiffen the fibrils and inhibit fibrillar sliding90,99. Collagen fibrils are stabilized by enzymatic and non-enzymatic crosslinks that form within and between fibrils100–103 (Fig. 1a). Whereas the rate of enzymatic crosslinking stabilizes at adolescence104,105, non-enzymatic crosslinks form the so-called advanced glycation end products (AGEs), which accumulate with age by up to five times the amount present in young bone90, 100,105–107. AGEs accumulate over time in association with reduced bone turnover, oxidative stress and dietary glucose intake108. AGE levels thus represent a surrogate marker for the age of bone extracellular matrix. AGEs also have the capacity to impact cellular function, including that of osteoblasts and osteoclasts108–110.
Accordingly, increased non-enzymatic crosslinking in age-ing cortical bone has been associated with diminished post-yield mechanical properties (Fig. 1b), ductility, toughness and fatigue resistance, both in human and animal models99,111–115. In the porous cancellous bone found within the extremities of long bones, AGEs have been shown to accumulate within the centre of the trabecular struts, where there is less resistance to fatigue crack expansion116. The role of crosslinking on ductility and toughness seems con-sistent with the bone fragility associated with certain diseases. In type 1 diabetes117 and in diabetic obesity, exposure to high levels of blood glucose boosts the formation of AGEs118. Under long-term antiresorptive treatments, renewal of bone matrix is hampered, allowing AGE crosslinking to accumulate29,119 (Fig. 1c). Similar elevations in the proportion of AGE crosslinks have been observed after irradiation120 (Fig. 1d).
Therefore, AGEs play a causal role in compromising the material quality of collagen itself. We believe that excessive non-enzymatic crosslinking reduces the critical crack size required for overload fracture by lowering the fracture toughness Kc in stage III of the fatigue process90. When the crack reaches this size, it can no lon-ger propagate progressively in fatigue mode, and the remaining
cross-section will break in a sudden and brittle way in overload mode. As such, a high density of AGEs is likely to allow an incipient fatigue crack to cause complete fracture of the bone prematurely.
Consequently, collagen crosslinking may provide a practical marker for the prediction of fracture toughness and of fatigue prop-erties independently of bone mass, both because of its direct role in compromising collagen behaviour, and of its indirect association with bone age90, 100,105–107. There is some promise that levels of pento-sidine (a marker for non-enzymatic crosslinking) in serum or urine is related to the levels of pentosidine in bone121. If confirmed, this could provide a simple test for predicting fracture risk associated with non-enzymatic collagen crosslinking. However, more stud-ies are warranted to investigate the possible relationship between fatigue fracture behaviour and the accumulation of non-enzymatic collagen crosslinks in bone. An improved understanding of colla-gen crosslinking and fatigue is needed to advance the development
of new therapies to restore bone quality and that complement the treatment of low bone mass.
Cellular mechanisms of damage repair
Bone stress fractures—that is, both insufficiency fractures and fatigue fractures—cannot only be studied from a purely mechanical or material perspective. Cortical bone undergoes fatigue damage as a result of cyclic loading at physiological stress and strain levels, usually under 0.2% strain9,10. At these strain levels, bone samples can sustain millions of cycles before failure when tested in vitro (that is, in cadav-eric bones)11. However, fractures occur after a few hundred thousand cycles when applied in vivo11, indicating the need to perform in vivo experiments, as in the recently published studies with rodent mod-els122–124. What is surprising is that the bone repair processes, which are active in vivo, would be expected to remove damage and inhibit or delay stress fractures (in comparison to in vitro tests); but this is 0.0 0.5 1.0 1.5 2.0 2.5 3.0 0.8 0.6 0.4 0.2 0 Young Aged Applied tissue strain 0.0 0.5 1.0 1.5 2.0 2.0 1.8 1.6 1.4 1.2 1.0 0.8 0.6 0.4 0.2 0 Irradiated bone 70 kGy Unirradiated bone
Whole bone strain (%) Whole bone strain (%)
Whole bone strain (%)
Collagen fibril strain (%)
Age Irradiation
b c BP treatment d
Non-enzymatic crosslinks
AGEs
(ng quinine per mg collagen)
1,000 800 600 400 200 0 50 40 30 20 10 0 180 160 140 120 100 80 60 40 20 0 Irradiated bone 70 kGy Unirradiated bone Treated Control Aged Young * 0 0.2 0.4 0.6 0.8 1.0 1.2 1.4 0 0.2 0.4 0.6 0.8 1.0 1.2 1.4 BP treated Control Haversian lamellae Haversian canals Cement lines Interstitial lamellae a
Mature enzymatic crosslinks (interfibrillar) Non-enzymatic crosslinks
(intra- and inter-fibrillar)
Fibre arrays ~ 5 μm
Secondary osteons ~ 100 μm Mineralized collagen fibrils
~ 1 μm Collagen molecules ~ 300 μm Immature enzymatic crosslinks (intermolecular) Hydroxyapatite nanocrystals ~ 10 μm Whole bone
Fig. 1 |Non-enzymatic crosslinking stiffens collagen fibrils.a, The multiscale structure of bone. b–d, The correlation between the accumulation of advanced glycation end product (AGE) crosslinks and diminished collagen fibril deformation in aged bone (b), in bisphosphonate (BP)-treated bone (c) and in X-ray-radiated bone (d). Top panels show data collected from in situ uniaxial tests performed in an X-ray synchrotron where small-angle X-ray scattering is used to reveal the actual strain carried by the collagen as compared to the strain in the whole bone tissue. Bottom panels show the proportion of non-enzymatic collagen crosslinks (AGEs) measured using a fluorometric assay29, 90,120. Error bars: b,d, standard deviation; c, standard error of the mean.
not always the case. This has been explained by means of feedback loops that can either promote bone damage or bone repair125. When bone resorption exceeds bone formation, the rate of damage forma-tion increases125. This scenario creates a positive feedback loop that boosts the amount of damage. This simplified explanation of damage repair mechanisms is, however, not fully satisfying because it does not take into account the timing of the repair process nor cell signal-ling. Here, we use ‘repair cycle’ to describe the process by which the damage is repaired normally, as opposed to ‘damage cycle’, where the repair cycle is interrupted because loading persists during the tran-sition between resorption and formation, or because fatigue crack formation and growth is faster than the remodelling rate (Fig. 2). However, the specific conditions in which in vivo fatigue loading can accelerate the occurrence of fatigue failure and the mechanisms responsible for this damage cycle remain unclear.
To consider the biological mechanisms that either prevent or pro-mote fatigue damage, it is worth briefly discussing the basic process of remodelling in healthy and fragile bone. Modelling refers to bone-forming osteoblasts depositing new bone at skeletal sites exposed to increased stress or strain and resulting in an updated bone shape, whereas remodelling involves bone resorption by osteoclasts, fol-lowed asynchronously by osteoblastic bone formation. Bone adap-tation via modelling and remodelling targets, at least partially, the repair of damaged regions54. In the elderly and in several conditions associated with clinical bone fragility, the rate of bone remodelling is low compared to that of young healthy bone. In addition, osteoclast activity can be increased compared to osteoblast activity, which often leads to loss of bone mass. These conditions are thought to create two cumulative detrimental consequences. First, the low bone mass induces higher levels of cyclic stress, which allows microdamage to initiate and grow more easily. Second, the reduced remodelling rate, along with the increased resorption porosities around microdamage, stimulates the damage cycle and thus rapid growth of fatigue dam-age. Moreover, bone quality is reduced with increasing age, in part because of a reduction in collagen plasticity90.
Osteocytes, which comprise 90% of the bone cell population, are major coordinators of bone remodelling. Embedded within the bone matrix, osteocytes are central to bone mechanotransduction
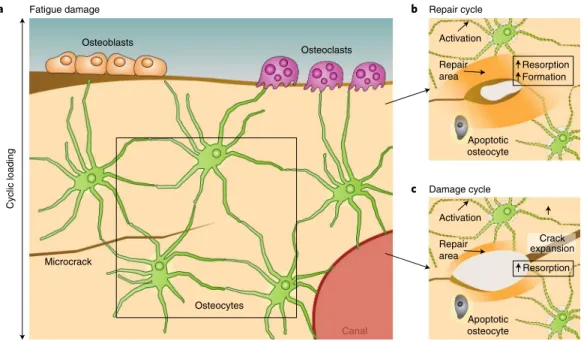
(the bone’s ability to sense and respond to mechanical stimuli126,127). Osteocytes extend tiny dendrite-like processes through canaliculi (microscopic channels between the lacunae of ossified bone) to communicate with neighbouring osteocytes, osteoblasts, osteo-clasts and blood vessels (Fig. 2a). Canaliculi are thought to sense changes in load and fatigue microdamage that induce resorption by osteoclasts56,128–130. Fatigue microcracks, by severing the cana-liculi, induce the local death of osteocytes through apoptosis11, 23,131. Osteocyte apoptosis then triggers the surrounding, surviving osteo-cytes to express RANKL131, a cytokine that promotes osteoclastic bone resorption132. This local increase in bone resorption removes microdamage and is normally followed by bone formation in the repair cycle. Yet if excessive loading persists during the transi-tion between resorptransi-tion and formatransi-tion, resorptransi-tion porosities can increase the local strains and accelerate the fracture process through the damage cycle11,125 (Fig. 2). On the contrary, diffuse damage does not seem to affect osteocyte viability nor trigger osteoclastic activity, but self-repairs over time23, perhaps via osteocyte-mediated perila-cunar remodelling. Indeed, osteocytes not only supervise osteoclast and osteoblast function, but also actively resorb and replace their surrounding bone matrix through perilacunar remodelling95,133–136. Osteocyte-mediated perilacunar remodelling plays an essential role in the biological control of bone quality. But the extent to which damage repair is executed directly by osteoclasts and by osteocytes remains an area of active investigation.
Between birth and the age of 80, the percentage of apoptotic osteocytes increases to up to ~45% in cortical bone137,138. Age-related osteocyte apoptosis is thought to be related to the deterioration in the canalicular network, which causes oxygen and nutrient depri-vation139. Glucocorticoid treatment and diseases (such as osteo-porosis, osteoarthritis and osteonecrosis) are also associated with elevated osteocytic apoptosis136, 140,141. Decreases in the number of viable osteocytes and canaliculi limit both the ability of osteocytes to sense microdamage and the release of RANKL to recruit osteo-clasts to repair it136, 142,143. Therefore, deficits in osteocyte function contribute in many ways to increased microcrack accumulation143 and to reduced fatigue resistance in the elderly. Moreover, osteocyte lacunae are subject to hypermineralization144, which may provide Osteoblasts Osteoclasts Osteocytes Canal Microcrack Damage cycle Repair cycle b c Fatigue damage a Apoptotic osteocyte Activation Repair area Resorption Formation Apoptotic osteocyte Resorption Activation Repair area Crack expansion Cyclic loading
Fig. 2 |Repair or acceleration of the formation of fatigue cracks.a, Bone cells in bone damaged by a microcrack. b, Repair cycle: the crack is repaired by resorption, followed by deposition of new bone. c, Damage cycle: the repair cycle is challenged or interrupted in its resorption phase, leading to stress concentration and to the expansion of existing cracks.
a preferential crack path for fatigue crack growth, analogously to crack growth preceding bone fracture under monotonic loading145.
Insufficiency fractures are believed to result from a high rate of fatigue-damage formation that exceeds the rate of immediate bone remodelling and repair125. The kinetics of fatigue damage and of damage repair, although not fully understood, dictate the fate of a loaded bone. Many questions remain about the timing of each stage of the repair process, the precise role of each cell type throughout the repair process, and the influence of fatigue-loading frequency and the recovery time between loadings. This highlights the impor-tance of carrying out in vivo fatigue studies to ascertain the compet-ing roles of fatigue damage and cellular repair.
Furthermore, we believe that it is crucial to elucidate the bio-logical pathways involved in fatigue damage repair. How do remod-elling factors differ from those involved in single-load-induced fracture repair? Several factors are induced in rat models of stress fracture; however, few of these (in particular, nitric oxide, sclerostin and prostaglandins, which are associated with bone remodelling136) have been functionally implicated in the response to fatigue dam-age. Future work will need to investigate the conditions by which fatigue can boost damage repair to prevent stress fracture. Although mice provide a powerful genetic model system, rabbit models may be more useful, as the Haversian organization of rabbit bone more closely resembles that of human bone146. Understanding these con-ditions may also help elucidate the mechanisms that promote stress fracture, and will hopefully guide the development of therapies capable of targeting each component of the repair cycle, including osteoblasts, osteoclasts and the direct and indirect actions of osteo-cytes. Combined with exercise, such pathways may be targeted to prevent insufficiency fractures and to maintain bone quality over nanometric to macrometric length scales in the ageing skeleton.
Prevention and monitoring of insufficiency fractures
There are two ways to combat insufficiency fractures associated with bone fragility: either preventing them altogether, or detecting, treating and monitoring them.Prevention. Because an insufficiency fracture is the result of an imbalance between cyclic load and damage repair, preventive pro-grams should focus on restoring the balance between loading and repair. Physically active elderly people are less prone to getting insufficiency fractures147–149. Therefore, guidance is needed to help calibrate a healthy duration and intensity of activity-related load-ing to the body’s capacity for repair. Although much remains to be learned, this can already be achieved in a number of ways. For example, the level of applied stress can be limited by using shock-absorbing shoes on softer surfaces150 and by reducing the intensity and duration of sport-led repeated loading to allow time for bone repair. Nutritional supplements, namely vitamin D and calcium, are highly recommended to prevent bone fracture151,152. Indeed, it has been recently shown that vitamin D plays a role in the regulation of osteocyte survival and in perilacunar remodelling153, which may be pivotal for its capacity to improve bone quality. Preventative steps can also minimize the skeletal impact of medical conditions that necessitate bed rest. Bedridden elderly people quickly lose muscle strength and bone mass.
Detection, monitoring and risk assessment. Following the lessons learned from fracture mechanics with composite materials, a ‘dam-age tolerant’ approach could be opted for; that is, the monitoring of damage accumulation and the limiting of critical damage growth instead of the prevention of damage formation. On the basis of the level of detected fatigue damage, the risk of fatigue-induced fracture can be assessed and repair actions taken. In structural engineering, most crack detection is performed by visual inspection of critical sites, aided by detection techniques such as alternating current or
ultrasonic testing. Ideally, in vivo fatigue cracking would be moni-tored on a yearly basis using radiation-free and affordable imaging that achieves the sensitivity currently available only with MRI or high-resolution computed tomography. An optical sensor consist-ing of a laser and a camera that can affordably distconsist-inguish between broken and unbroken bones without radiation154 shows promise, but this technique has not yet been tested on insufficiency fractures.
Another approach for assessing in vivo fracture risk would be to measure the critical fracture toughness, which defines the transition from progressive crack growth to catastrophic failure. To that end, since the early 1900s, hardness indentation tests have been used to quantify hardness and elastic modulus. The osteopenetrometer, developed in the 1980s, was based on this technique to measure tra-becular bone strength during surgical procedures155. More recently, reference point indentation (RPI), an in vivo microindentation tech-nique, has been shown to collect outcomes that (in its first version) correlate with a reduction in bone toughness21,156–158. In studies on aged patients156, the increase in indentation distance of the RPI was found to correlate negatively with crack-growth toughness (patients with osteoporotic fractures had a 47% greater increase in indenta-tion distance compared to age-matched controls). A newer version of the RPI159,160 was designed as a single-impulse indentation, which resembles more that of impact testers (such as the Charpy test devel-oped in the early 1900s). Impact tests have been used to measure the energy absorbed during dynamic fracture and specifically to study the ductile–brittle transition. Although the outcomes of the newer RPI have not yet been compared with traditional mechanical prop-erties159,160, they should mainly reflect pre-yield properties, because post-yield and extrinsic toughening mechanisms are diminished in bone at high strain rates161,162.
Although these techniques appear to be promising as clinical measurements of fracture risk in living patients, the mechanisms that they are investigating have not yet been shown to correlate with fatigue fracture mechanisms. Furthermore, these measures are lim-ited to a local region of the tibial cortical bone, yet are performed as a blind test with the nominal assumption of homogeneous mechan-ical properties. Moreover, the use of these techniques is question-able on aged bone, where microindenting and impacting could be the loading event that triggers fracture of bone weakened by fatigue damage accumulation.
Levels of non-enzymatic collagen crosslinking may also pro-vide a practical marker to predict fracture risk. Other biochemical markers (such as oestradiol, dehydroepiandrosterone sulfate, sex-hormone-binding globulin and parathyroid hormone) might also be an effective means to monitor bone remodelling and metabolism and to evaluate the risk of insufficiency fracture163.
The future of the prevention of insufficiency fractures may involve all these parameters and rely on the use of health monitoring systems. Simple bone health parameters such as vital signs, weight, age, levels of activity (including exercise, bed-rest time and the pres-ence of extreme loadings), food intake and nutritional supplement intake, as well as medical information such as that from bone imag-ing, bone densitometry, in vivo microindentation outcomes, blood tests and treatments, could be monitored. Statistical analysis of sen-sitive parameters would then provide a prediction of the patient’s bone health as well as key strategies for maintaining bone integrity.
Outlook
The interactions between cyclic loading, fatigue damage and repair processes are complex. In this Perspective, we have provided evi-dence that suggests that insufficiency fractures in the elderly pop-ulation might be the result of an imbalance between damage and repair dynamics that are highly sensitive to changes in bone quality, bone mass and remodelling with age. To unravel the complexities, we are proposing to approach this problem from a somewhat differ-ent viewpoint. On the basis of case studies, we postulate that bone
fractures in the elderly, perhaps for a quarter of them, are insuf-ficiency fractures caused by cyclic loads inducing fatigue crack-ing, and not necessarily following impacts or trauma. We have also presented the concepts of fatigue crack growth, inherited from the fracture mechanics of non-biological materials, and discussed experimental clues that indicate that bone might have similar fatigue behaviour in vitro. We believe that it is imperative to investigate the deterioration in bone quality in aged bones. The loss of collagen plasticity, combined with increased osteon density, plays important roles in the increase of microdamage density with age. This might explain how an incipient fatigue crack can cause the complete frac-ture of aged bone premafrac-turely.
Cellular mechanisms act to repair fatigue damage in vivo and prevent stress fractures (the repair cycle). However, in ageing or in conditions of bone fragility, the timing of repair is challenged by excessive rates of fatigue damage formation or by low turnover rate. In these circumstances, the cellular mechanisms of repair are insuf-ficient and the fatigue damage propagation is accelerated (the dam-age cycle). The biological pathways and the time-dependencies of the repair and damage cycles still need to be elucidated. Osteocytes are already known to be involved in the activation of bone resorp-tion and formaresorp-tion around damage. New evidence indicates that they are also actors in the local remodelling process. Nonetheless, there are still major gaps in the understanding of the role of osteo-cytes in damage repair. Future clinical indicators of fracture risk will be based on bone quality, such as serum or urine markers of bone remodelling or collagen crosslinking, on new imaging modalities, or on in vivo microindentation techniques, instead of relying solely on bone mass. And new therapeutic treatments that stimulate the cellular repair of fatigue damage may restore bone quality, or at least prevent its degeneration.
Received: 19 April 2017; Accepted: 7 December 2017; Published online: 22 January 2018
References
1. Cole, J. H. & van der Meulen, M. C. Whole bone mechanics and bone quality. Clin. Orthopaed. Rel. Res. 469, 2139–2149 (2011).
2. Breithaupt, J. Zur pathologie des menschlichen fusses. Med. Zeitung 24, 169–171 (1855).
3. Pirker, H. Bruch der oberschenkeldiaphyse durch muskelzug. Arch. Klin. Chir. 175, 155–168 (1934).
4. Burrows, H. J. Spontaneous fracture of the apparently normal fibula in its lowest third. British J. Surg. 28, 82–87 (1940).
5. Fredericson, M., Jennings, F., Beaulieu, C. & Matheson, G. O. Stress fractures in athletes. Topics in Magnetic Resonance Imaging 17, 309–325 (2006).
6. Brukner, P., Bradshaw, C. & Bennell, K. Managing common stress fractures: let risk level guide treatment. Phys. Sports Med. 26, 39–47 (1998). 7. Iwamoto, J. & Takeda, T. Stress fractures in athletes: review of 196 cases.
J. Orthopaed. Sci. 8, 273–278 (2003).
8. Meurman, K. & Elfving, S. Stress fracture in soldiers: a multifocal bone disorder. A comparative radiological and scintigraphic study. Radiology.
134, 483–487 (1980).
9. Schaffler, M., Radin, E. & Burr, D. Long-term fatigue behavior of compact bone at low strain magnitude and rate. Bone. 11, 321–326 (1990). 10. Schaffler, M., Radin, E. & Burr, D. Mechanical and morphological effects of
strain rate on fatigue of compact bone. Bone. 10, 207–214 (1989). 11. Schaffler, M. B. in Musculoskeletal Fatigue and Stress Fractures 161–182
(CRC, Boca Raton, 2000).
12. Pentecost, R. L., Murray, R. A. & Brindley, H. H. Fatigue, insufficiency, and pathologic fractures. JAMA 187, 1001–1004 (1964).
13. Breer, S. et al. Stress fractures in elderly patients. Int. Orthopaed. 36, 2581–2587 (2012).
14. Carpintero, P., Berral, F. J., Baena, P., Garcia-Frasquet, A. & Lancho, J. L. Delayed diagnosis of fatigue fractures in the elderly. Am. J. Sports Med. 25, 659–662 (1997).
15. Miller, K. E. Diagnosis of insufficiency fracture in the elderly. Am. Fam. Phys. 57, 1968–1968 (1998).
16. Kaye, R. A. Insufficiency stress fractures of the foot and ankle in postmenopausal women. Foot Ankle Int. 19, 221–224 (1998).
17. Bentolila, V. et al. Intracortical remodeling in adult rat long bones after fatigue loading. Bone 23, 275–281 (1998).
18. Colopy, S. et al. Response of the osteocyte syncytium adjacent to and distant from linear microcracks during adaptation to cyclic fatigue loading.
Bone 35, 881–891 (2004).
19. Frost, H. Presence of microscopic cracks in vivo in bone. Henry Ford Hosp. Med. Bull. 8, 35 (1960).
20. Burr, D. B. et al. Bone microdamage and skeletal fragility in osteoporotic and stress fractures. J. Bone Miner. Res. 12, 6–15 (1997).
21. Nyman, J. S. & Makowski, A. J. The contribution of the extracellular matrix to the fracture resistance of bone. Curr. Osteopor. Rep. 10, 169–177 (2012). 22. Jepsen, K. J. The aging cortex: to crack or not to crack. Osteopor. Int. 14,
57–66 (2003).
23. Seref-Ferlengez, Z., Kennedy, O. D. & Schaffler, M. B. Bone microdamage, remodeling and bone fragility: how much damage is too much damage?
BoneKEy Rep. 4, 644 (2015).
24. Hernandez, C. J. Bone fatigue, stress fractures and bone repair (Sun Valley 2013). BoneKEy Rep. 10, 448 (2013).
25. Krestan, C. & Hojreh, A. Imaging of insufficiency fractures. Eur. J. Radiol.
71, 398–405 (2009).
26. Lenart, B. et al. Association of low-energy femoral fractures with pro- longed bisphosphonate use: a case control study. Osteopor. Int. 20, 1353–1362 (2009).
27. Ettinger, B., Burr, D. B. & Ritchie, R. O. Proposed pathogenesis for atypical femoral fractures: lessons from materials research. Bone 55, 495–500 (2013). 28. Shane, E. et al. Atypical subtrochanteric and diaphyseal femoral fractures:
report of a task force of the american society for bone and mineral research. J. Bone Miner. Res. 25, 2267–2294 (2010).
29. Acevedo, C. et al. Alendronate treatment alters bone tissues at multiple structural levels in healthy canine cortical bone. Bone 81, 352–363 (2015). 30. Aspenberg, P. Atypical fractures, a biased perspective. Injury. 47, S28–S30
(2016).
31. Mann, G. et al. in Sports Injuries 787–813 (Springer, Berlin, 2012). 32. Soubrier, M. et al. Insufficiency fracture: a survey of 60 cases and review of
the literature. Joint Bone Spine 70, 209–218 (2003).
33. Frey, M. E. et al. Percutaneous sacroplasty for osteoporotic sacral insufficiency fractures: a prospective, multicenter, observational pilot study.
Spine J. 8, 367–373 (2008).
34. Moran, D. S., Evans, R. K. & Hadad, E. Imaging of lower extremity stress fracture injuries. Sports Med. 38, 345–356 (2008).
35. Idiyatullin, D., Garwood, M., Gaalaas, L. & Nixdorf, D. R. Role of MRI for detecting micro cracks in teeth. Dentomaxillofac. Radiol. 45, 20160150 (2016). 36. Zimmermann, E. A., Busse, B. & Ritchie, R. O. The fracture mechanics of
human bone: influence of disease and treatment. BoneKEy Rep. 4, 743 (2015).
37. Hernandez, C. J. & van der Meulen, M. C. Understanding bone strength is not enough. J. Bone Miner. Res. 32, 1157–1162 (2017).
38. Fujiwara, S. Importance of raising awareness about spontaneous insufficiency fractures in the bedridden elderly. Int. J. Clin. Rheumatol. 5, 395–397 (2010).
39. Takamoto, S. et al. Spontaneous fractures of long bones associated with joint contractures in bedridden elderly inpatients: clinical features and outcome. J. Am. Geriatr. Soc. 53, 1439–1441 (2005).
40. Taillandier, J., Langue, F., Alemanni, M. & Taillandier-Heriche, E. Mortality and functional outcomes of pelvic insufficiency fractures in older patients.
Joint Bone Spine. 70, 287–289 (2003).
41. Schwendner, K. I., Mikesky, A. E., Holt, W. S. Jr, Peacock, M. & Burr, D. B. Differences in muscle endurance and recovery between fallers and nonfallers, and between young and older women. J. Gerontol. Ser. A Biol. Sci. Med. Sci 52, M155–M160 (1997).
42. Burr, D. B. Muscle strength, bone mass, and age-related bone loss. J. Bone Miner. Res. 12, 1547–1551 (1997).
43. Sloan, J. & Holloway, G. Fractured neck of the femur: the cause of the fall?
Injury 13, 230–232 (1981).
44. Dorne, H. & Lander, P. H. Spontaneous stress fractures of the femoral neck.
Am. J. Roentgenol. 144, 343–347 (1985).
45. Tountas, A. A. Insufficiency stress fractures of the femoral neck in elderly women. Clin. Orthopaed. Rel. Res. 292, 202–209 (1993).
46. Lyders, E., Whitlow, C., Baker, M. & Morris, P. Imaging and treatment of sacral insufficiency fractures. Am. J. Neuroradiol. 31, 201–210 (2010). 47. Weber, M., Hasler, P. & Gerber, H. Insufficiency fractures of the sacrum:
twenty cases and review of the literature. Spine 18, 2507–2512 (1993). 48. West, S. G., Troutner, J. L., Baker, M. R. & Place, H. M. Sacral insufficiency
fractures in rheumatoid arthritis. Spine 19, 2117–2121 (1994). 49. Featherstone, T. Magnetic resonance imaging in the diagnosis of sacral
stress fracture. British J. Sports Med. 33, 276–277 (1999).
50. Kane, R. S., Burns, E. A. & Goodwin, J. S. Minimal trauma fractures in older nursing home residents: the interaction of functional status, trauma, and site of fracture. J. Am. Geriatr. Soc. 43, 156–159 (1995).
51. Lapina, O. & Tiskevicius, S. Sacral insufficiency fracture after pelvic radiotherapy: a diagnostic challenge for a radiologist. Medicina 50, 249–254 (2014).
52. Haentjens, P. et al. Meta-analysis: excess mortality after hip fracture among older women and men. Ann. Intern. Med. 152, 380–390 (2010).
53. LeBlanc, E. S. et al. Hip fracture and increased short-term but not long-term mortality in healthy older women. Arch. Intern. Med. 171, 1831–1837 (2011).
54. Taylor, D., Hazenberg, J. G. & Lee, T. C. Living with cracks: damage and repair in human bone. Nat. Mater. 6, 263–268 (2007).
55. Poundarik, A. A. et al. Dilatational band formation in bone. Proc. Natl Acad. Sci. USA 109, 19178–19183 (2012).
56. Mori, S. & Burr, D. Increased intracortical remodeling following fatigue damage. Bone 14, 103–109 (1993).
57. Duncan, R. & Turner, C. Mechanotransduction and the functional response of bone to mechanical strain. Calc. Tissue Int. 57, 344–358 (1995). 58. Lanyon, L. E. & Rubin, C. Static vs dynamic loads as an influence on bone
remodelling. J. Biomech. 17, 897–905 (1984).
59. Carter, D., Blenman, P. & Beaupre, G. Correlations between mechanical stress history and tissue differentiation in initial fracture healing. J. Orthopaed. Res. 6, 736–748 (1988).
60. Ehrlich, P. & Lanyon, L. Mechanical strain and bone cell function: a review.
Osteopor. Int. 13, 688–700 (2002).
61. Diab, T., Sit, S., Kim, D., Rho, J. & Vashishth, D. Age-dependent fatigue behaviour of human cortical bone. Eur. J. Morphol. 42, 53–59 (2005). 62. Diab, T., Condon, K. W., Burr, D. B. & Vashishth, D. Age-related change in
the damage morphology of human cortical bone and its role in bone fragility. Bone 38, 427–431 (2006).
63. Diab, T. & Vashishth, D. Morphology, localization and accumulation of in vivo microdamage in human cortical bone. Bone 40, 612–618 (2007). 64. Schaffler, M., Choi, K. & Milgrom, C. Aging and matrix microdamage
accumulation in human compact bone. Bone 17, 521–525 (1995). 65. Zioupos, P., Gresle, M. & Winwood, K. Fatigue strength of human cortical
bone: age, physical, and material heterogeneity effects. J. Biomed. Mater. Res. 86, 627–636 (2008).
66. Norman, T. L. & Wang, Z. Microdamage of human cortical bone: incidence and morphology in long bones. Bone 20, 375–379 (1997).
67. Laird, C. & Smith, G. Crack propagation in high stress fatigue. Phil. Mag. 7, 847–857 (1962).
68. Suresh, S. Fatigue of Materials (Cambridge Univ. Press, Cambridge, 1998). 69. Anderson, T. L. Fracture Mechanics: Fundamentals and Applications (CRC,
Boca Raton, 2005).
70. Broek, D. Elementary Engineering Fracture Mechanics (Springer Science & Business Media, 2012).
71. Robertson, S. W. & Ritchie, R. O. In vitro fatigue–crack growth and fracture toughness behavior of thin-walled superelastic nitinol tube for endovascular stents: a basis for defining the effect of crack-like defects.
Biomaterials 28, 700–709 (2007).
72. Zwas, S. T., Elkanovitch, R. & Frank, G. Interpretation and classification of bone scintigraphic findings in stress fractures. J. Nucl. Med. 28, 452–457 (1987).
73. Milgrom, C. et al. Multiple stress fractures: A longitudinal study of a soldier with 13 lesions. Clin. Orthopaed. Rel. Res. 192, 174–179 (1985).
74. Carter, D. & Caler, W. A cumulative damage model for bone fracture. J. Orthopaed. Res. 3, 84–90 (1985).
75. Nalla, R. K., Kruzic, J. J., Kinney, J. H. & Ritchie, R. O. Aspects of in vitro fatigue in human cortical bone: time and cycle dependent crack growth.
Biomaterials 26, 2183–2195 (2005).
76. Ritchie, R. O., Kinney, J. H., Kruzic, J. J. & Nalla, R. K. A fracture mechanics and mechanistic approach to the failure of cortical bone. Fatig. Fract. Eng. Mater. Struct. 28, 345–371 (2005).
77. Paris, P. C. & Erdogan, F. A critical analysis of crack propagation laws. J. Basic Eng. 528–534 (1963).
78. Pattin, C., Caler, W. & Carter, D. Cyclic mechanical property degradation during fatigue loading of cortical bone. J. Biomech. 29, 69–79 (1996). 79. Genant, H. K. et al. Interim report and recommendations of the world
health organization task-force for osteoporosis. Osteopor. Int. 10, 259–264 (1999).
80. Schuit, S. et al. Fracture incidence and association with bone mineral density in elderly men and women: the Rotterdam study. Bone 34, 195–202 (2004).
81. Hui, S. L., Slemenda, C. W. & Johnston, C. C. Jr Age and bone mass as predictors of fracture in a prospective study. J. Clin. Invest. 81, 1804 (1988).
82. Allolio, B. Risk factors for hip fracture not related to bone mass and their therapeutic implications. Osteopor. Int. 9, S9–S17 (1999).
83. Sandor, T., Felsenberg, D. & Brown, E. Comments on the hypotheses underlying fracture risk assessment in osteoporosis as proposed by the world health organization. Calc. Tissue Int. 64, 267–270 (1999).
84. Mccreadie, B. R. & Goldstein, S. A. Biomechanics of fracture: is bone mineral density sufficient to assess risk? J. Bone Miner. Res. 15, 2305–2308 (2000).
85. Heaney, R. P. Is the paradigm shifting? Bone 33, 457–465 (2003).
86. Ritchie, R. O., Buehler, M. J. & Hansma, P. Plasticity and toughness in bone.
Phys. Today 62, 41–47 (2009).
87. Zioupos, P. & Currey, J. Changes in the stiffness, strength, and toughness of human cortical bone with age. Bone 22, 57–66 (1998).
88. McCalden, R., McGeough, J. & Barker, M. et al. Age-related changes in the tensile properties of cortical bone: the relative importance of changes in porosity, mineralization, and microstructure. JBJS 75, 1193–1205 (1993). 89. Burstein, A. H., Reilly, D. T. & Martens, M. Aging of bone tissue:
mechanical properties. JBJS 58, 82–86 (1976).
90. Zimmermann, E. A. et al. Age-related changes in the plasticity and toughness of human cortical bone at multiple length scales. Proc. Natl Acad. Sci. USA 108, 14416–14421 (2011).
91. Launey, M. E., Buehler, M. J. & Ritchie, R. O. On the mechanistic origins of toughness in bone. Annu. Rev. Mater. Res. 40, 25–53 (2010).
92. Eppell, S. J., Smith, B., Kahn, H. & Ballarini, R. Nano measurements with micro-devices: mechanical properties of hydrated collagen fibrils. J. R. Soc. Interf. 3, 117–121 (2006).
93. Ker, R. in Collagen: Structure and Mechanics 111–131 (Springer, New York, 2008).
94. Manhard, M. K. et al. MRI-derived bound and pore water concentrations as predictors of fracture resistance. Bone 87, 1–10 (2016).
95. Tang, S. Y., Herber, R.-P., Ho, S. P. & Alliston, T. Matrix
metalloproteinase–13 is required for osteocytic perilacunar remodeling and maintains bone fracture resistance. J. Bone Miner. Res. 27, 1936–1950 (2012).
96. Thurner, P. J. et al. Osteopontin deficiency increases bone fragility but preserves bone mass. Bone 46, 1564–1573 (2010).
97. Boskey, A., DiCarlo, E., Paschalis, E., West, P. & Mendelsohn, R. Comparison of mineral quality and quantity in iliac crest biopsies from highand low-turnover osteoporosis: an FT-IR microspectroscopic investigation. Osteopor. Int. 16, 2031–2038 (2005).
98. Alliston, T. Biological regulation of bone quality. Curr. Osteopor. Rep. 12, 366–375 (2014).
99. Vashishth, D. et al. Influence of nonenzymatic glycation on biomechanical properties of cortical bone. Bone 28, 195–201 (2001).
100. Sell, D. R. & Monnier, V. Structure elucidation of a senescence crosslink from human extracellular matrix. implication of pentoses in the aging process. J. Biol. Chem. 264, 21597–21602 (1989).
101. Bailey, A. J. Molecular mechanisms of ageing in connective tissues. Mech. Aging Dev. 122, 735–755 (2001).
102. Boskey, A. L. & Coleman, R. Aging and bone. J. Dent. Res. 89, 1333–1348 (2010).
103. Saito, M. & Marumo, K. Collagen cross-links as a determinant of bone quality: a possible explanation for bone fragility in aging, osteoporosis, and diabetes mellitus. Osteopor. Int. 21, 195–214 (2010).
104. Eyre, D. R., Dickson, I. & Van Ness, K. Collagen cross-linking in human bone and articular cartilage. age-related changes in the content of mature hydroxypyridinium residues. Biochem. J. 252, 495–500 (1988).
105. Saito, M., Marumo, K., Fujii, K. & Ishioka, N. Single-column high-performance liquid chromatographic-fluorescence detection of immature, mature, and senescent cross-links of collagen. Analyt. Biochem. 253, 26–32 (1997).
106. Nyman, J. S. et al. Age-related effect on the concentration of collagen crosslinks in human osteonal and interstitial bone tissue. Bone 39, 1210–1217 (2006).
107. Odetti, P. et al. Advanced glycation end products and bone loss during aging. Ann. NY Acad. Sci. 1043, 710–717 (2005).
108. Ott, C. et al. Role of advanced glycation end products in cellular signaling.
Redox Biol. 2, 411–429 (2014).
109. Zhou, Z. et al. Regulation of osteoclast function and bone mass by rage.
J. Exp. Med. 203, 1067–1080 (2006).
110. Miyata, T. et al. Advanced glycation end products enhance osteoclast-induced bone resorption in cultured mouse unfractionated bone cells and in rats implanted subcutaneously with devitalized bone particles. J. Am. Soc. Nephrol. 8, 260–270 (1997).
111. Wang, X., Shen, X., Li, X. & Agrawal, C. M. Age-related changes in the collagen network and toughness of bone. Bone 31, 1–7 (2002).
112. Garnero, P. et al. Extracellular post-translational modifications of collagen are major determinants of biomechanical properties of fetal bovine cortical bone. Bone 38, 300–309 (2006).
113. Siegmund, T., Allen, M. R. & Burr, D. B. Failure of mineralized collagen fibrils: modeling the role of collagen cross-linking. J. Biomech. 41, 1427–1435 (2008).
114. Tang, S. & Vashishth, D. Non-enzymatic glycation alters microdamage formation in human cancellous bone. Bone 46, 148–154 (2010).