SYNTHESIS AND ANTIMALARIAL EVALUATION OF A
QUINOLINE-TRIOXANE HYBRID DRUG
Wamakima Hannah Nyakio (B.Ed Sc.)
I56/22613/2011
A thesis submitted in partial fulfilment of the requirements for the award
of the Degree of Master of Science (Chemistry) in the School of Pure and
Applied Sciences of Kenyatta University
DECLARATION
I declare that this thesis is my original work and has not been presented for a degree in any other University or for any other award.
Signature ……… Date ………
Wamakima Hannah Nyakio
I56/22613/2011
Supervisors’ Approval
We confirm that the work reported in this thesis was carried out by the candidate under our supervision and has been submitted with our approval as university supervisors.
Signature ……… Date ………
Dr Margaret M. Ng‟ang‟a Department of Chemistry Kenyatta University
Signature ……… Date ………
Dr Peter G. Kirira
Department of Physical Sciences Mount Kenya University
Signature ……… Date ………
Dr Francis W. Muregi
DEDICATION
This thesis is dedicated to my husband, from whom I have learnt that pursuance of excellence is rewarding. It is also dedicated to my sons Eric and Bryan who were
ACKNOWLEDGEMENTS
My highest gratitude goes to God Almighty who has walked with me through this journey. He has given me grace, sustenance and comfort one day at a time.
I am deeply grateful to my supervisor Dr Peter G. Kirira, for opening my eyes to a new and very interesting field of research in hybrid drugs. I admire your scientific prowess and willingness to share your insights, skills and knowledge. I also greatly appreciate your flexibility and availability for consultation at all times, and the amount of time you spent painstakingly reading draft after draft of my work. Working on this project under your guidance has been a great learning experience. To my supervisor, Dr Margaret M. Ng‟ang‟a, thank you for making sure that everything was in order and diligently helping me to go through the thesis. Thank you for your useful suggestions and advice. I admire your ability to balance academic interests and personal pursuits. Above all, you made me feel a friend, which I appreciate from my heart.
My very heartfelt thanks go to the Acting Director, Centre for Traditional Medicine and Drug Research (CTMDR), Dr Jeniffer Orwa for giving me an opportunity to work in the CTMDR laboratory and all the staff for ensuring I had all I needed to successfully complete my research project. I am grateful to Mr Daniel Kiboi, for providing me with the parasites for this study, being there for consultation and for timely and helpful comments. Many thanks to Ms Beatrice Irungu, your high scientific standards, and hard work set me the tempo.
I dearly appreciate my love, Dr Francis W. Muregi, for being a wonderful friend and for introducing me to the world of research long before I started this study. Many are the nights you sat with me until the wee hours. The sacrifices did not go unnoticed. Your technical excellence and tremendous grasp of experimental issues was of great help to me.
Finally and not least, I greatly appreciate National Council of Science, Technology and Innovation which partially funded this work through Postgraduate Research Grant (Grant Ref. No.; NCST/ST&I/RCD/4thCall M.Sc/167, Appendix 1). Equally, I am very grateful to the Academy of Sciences for the Developing World (TWAS) whose grant to Dr Muregi (Grant Ref. No.; 11-082 RG/BIO/AF/AC_I; UNESCO FR: 3240262705) largely funded this work.
TABLE OF CONTENTS
DECLARATION ... ii
DEDICATION ... iii
ACKNOWLEDGEMENTS ... iv
TABLE OF CONTENTS ... v
LIST OF TABLES ... viii
LIST OF FIGURES ... iix
LIST OF SCHEMES ... iii
LIST OF APPENDICES ... x
ABBREVIATIONS AND ACRONYMS ... xi
CHAPTER ONE ... 1
INTRODUCTION ... 1
1.1 Background of the Study ... 1
1.2 Problem statement ... 3
1.3 Justification of the study ... 3
1.4 Hypothesis ... 4
1.5 Objectives ... 4
1.5.1 General objective ... 4
1.5.2 Specific objectives ... 4
CHAPTER TWO ... 6
LITERATURE REVIEW ... 6
2.1 Historical background of malaria ... 6
2.2 Disease transmission ... 8
2.3 Malaria chemotherapy ... 9
CHAPTER THREE ... 15
MATERIALS AND METHODS ... 15
3.1 Chemicals, glassware and study site ... 15
3.2 Synthetic design of quinoline-trioxane hybrid molecule ... 15
3.2.1 Introduction of a linker ... 16
3.2.2 Coupling procedure ... 16
3.3 General instrumentation ... 17
3.4 Bioassays of the hybrid drug ... 18
3.4.1 In vitro assays ... 18
3.4.1.1 Drug solutions ... 18
3.4.1.2 Plasmodium falciparum cultures ... 18
3.4.1.3 Evaluation of antiplasmodial activity of the hybrid drug ... 19
3.4.1.4 Combination studies of the individual precursors of the hybrid drug . 20 3.4.2. In vivo assays ... 20
3.4.2.1 Parasite, host and ethics ... 20
3.4.2.3 In vivo antiplasmodial evaluation of the hybrid drug ... 22
3.4.3 In vivo combination studies for the hybrid drug precursors ... 22
3.4.4 Studies of the hybrid drug against lumefantrine-resistant parasites ... 23
3.4.5 Cytotoxicity assays ... 23
3.5 Data analysis ... 25
CHAPTER FOUR ... 26
RESULTS AND DISCUSSION ... 26
4.1 Structure elucidation of 7-chloro-4-(1, 2-diaminoethyl)quinoline (16) ... 26
4.2 Structure elucidation of N-(7-chloroquinolin-4-ylamino) ethylartesunate 19- carboxamide (17)………... ... 28
4.3 Biological studies of the hybrid drug ... 34
4.3.1 In vitro studies ... 34
4.3.1.1 In vitro inhibition of Plasmodium growth by the hybrid drug ... 34
4.3.1.2 Potency of the hybrid drug and its precursors ... 36
4.3.2 In vivo studies ... 38
4.3.2.1 In vivo inhibition of Plasmodium growth by the hybrid drug... 38
4.3.2.2 In vivo potency of the hybrid drug and its precursors ... 41
4.3.3 Cytotoxicity studies ... 44
4.3.3.1 In vitro safety of the hybrid drug ... 44
CHAPTER FIVE ... 47
CONCLUSION AND RECOMMENDATIONS ... 47
5.1 Conclusion ... 47
5.2 Recommendations ... 48
REFERENCES ... 50
LIST OF TABLES
Table Title Page
Table 4.1 1H-NMR (200MHz) data of compound 16 in CDCI3………....…... 26 Table 4.2 13C-NMR (50MHz) data of compound 16 in CDCI3……… 28 Table 4.3 Percentage (%) yield of compound 17 using various coupling
reagents……….……. 29
Table 4.4 1H-NMR (200MHz) data of compound 17 in CDCI3………... 30 Table 4.5 13C-NMR (50MHz) data of compound 17 in CDI3………... 31 Table 4.6 IC50 of dual-drug, artesunate, chloroquine (CQ),
4,7-dichloroquinoline (DQ) against CQ-sensitive (D6) and -resistant
(W2) isolates………. 35
Table 4.7 In vitro inhibition (%) of parasite growth by the hybrid drug and precursors (artesunate and 4,7-dichloroquinoline) in
combination………... 36
Table 4.8 In vivo antiplasmodial activities of the hybrid drug, artesunate and 4,7-dichloroquine against P. berghei in mice determined
by the suppressive test in mice……….. 38
Table 4.9 In vivo antiplasmodial activity of artesunate (AS) and 4,7-dichloro- quinoline (DQ) drug combinations on day 4 and 7 pi against P. berghei ANKA determined by the suppressive test in
mice………... 42
Table 4.10 In vivo antiplasmodial activities of lumefantrine and the hybrid molecule on day 4 and 7 pi against P. berghei ANKA determined
by the suppressive test……….………. 43
Table 4.11 Percentage inhibition of Hep2 cell line by the dual drug,
artesunate, 4,7-dichloroquinoline and chloroquine………... 45
Table 4.12 IC50 values for the dual drug, artesunate, 4,7-dichloroquinoline and CQ against Hep2 cell line, CQR parasites and the selectivity
LIST OF FIGURES
Figure 2.1 Life cycle of malaria parasites………. 8
Figure 4.1 Correlations observed in H-H cosy of compound………..….. 27
Figure 4.2 Dose-activity relationship of the hybrid on day 4 and 7 post
infection……… 39
Figure 4.3 Effective dose-50 (ED50) and ED90 values for the hybrid drug……… 40 Figure 4.4 Percentage Hep2 cells viability grown in the presence of hybrid drug
LIST OF SCHEMES
Figure3.1 Synthetic strategy for hybrid molecule………..… 15
Figure3.2 Introduction of a linker……….. 16
LIST OF APPENDICES
Appendix1 Science, Technology & Innovation Research Grant Offer Letter… 56
Appendix2 KEMRI Ethics Review Committee Approval Certificate………… 57
Appendix3 IR spectrum of 7-chloro-4-(1,2-diaminoethyl)quinoline (16)…….. 58
Appendix3A 1H-NMR spectrum of 7-chloro-4-(1,2-diaminoethyl)quinoline
(16)………..… 59
Appendix3B 13C-NMR spectrum of 7-chloro-4-(1,2-diaminoethyl)quinolone
(16)………... 60
Appendix3C LC-MS spectrum of 7-chloro-4-(1,2-diaminoethyl)quinoline
(16)………... 61
Appendix 4 IR spectrum of N-(7-chloroquinolin-4-yamino)
ethyl-artesunate-19-carboxamide...………... 62
Appendix4A 1H-NMR spectrum of N-(7-chloroquinolin-4-yamino) ethyl-artesunate-19 carboxamide………...
63
Appendix4B 13C-NMR spectrum of N-(7-chloroquinolin-4-yamino)
ethyl-artesunate-19-carboxamide………... 64
Appendix4C LC-MS spectrum of N-(7-chloroquinolin-4-yamino)
ethyl-artesunate-19-carboxamide………... 65
Appendix4D LC-MS spectrum of N-(7-chloroquinolin-4-yamino)
ABBREVIATIONS AND ACRONYMS
ACT Artemisinin-based combination therapy AIDS Acquired immune deficiency syndrome
CDI Carbonyldiimidazole
COSY Correlation SpectroscopY
CTMDR Centre for Traditional Medicine and Drug Research
CQ Chloroquine
DEPT Distortionless Enhancement of Polarization Transfer DCC Dicyclohexylcarbodiimide
DIPEA Diisopropylethylamine DMAP 4-Dimethylaminopyridine
DV Digestive vacuole
ED50 Effective dose that reduces parasitaemia by 50%
EDC 1-Ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride HIV Human immunodeficiency virus
HOBt 1-Hydroxy-benzotriazole
KEMRI Kenya Medical Research Institute
PfCRT Plasmodium falciparum chloroquine resistance transporter PO Per os (oral administration)
PPM Parts per million
ABSTRACT
The global malaria situation is being exacerbated by emergence of drug resistance to conventional antimalarials, necessitating search for novel drugs. A recent rational approach of antimalarial drug design characterized as „„covalent bitherapy‟‟ involves linking two molecules with individual intrinsic activity into a single agent, thus packaging dual-activity into a single hybrid molecule. This study proposed to synthesize a dual drug based on quinoline and trioxane pharmacophoric scaffold of quinolines and artemisinins, respectively. The synthetic design involved introduction of a linker to 4,7-dichloroquinoline and subsequent coupling with artesunate to form the dual drug. The reactions were monitored by TLC and the compounds purified by recrystallisation and silica gel column. The yield for the dual drug was between trace and 71% depending on the coupling agent and solvent used. A combination of 1-Ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDC - coupling agent), 1-Hydroxy-benzotriazole (HOBt - additive) and dichloromethane (solvent) gave the highest yield of 71%. The chemical structures of the synthesized compounds were confirmed by 13C, and 1H-NMR and GC-MS studies. The dual drug was tested in vitro against chloroquine (CQ)-susceptible (CQS, D6) and-resistant (CQR, W2) Plasmodium falciparum strains. The 50% inhibitory concentration (IC50) values revealed that the drug was as active against both strains as artesunate (p>0.05) irrespective of the strain. The dual drug was more potent than CQ against both strains, with IC50 of 6.89 ηg ml-1 for the CQ-sensitive (CQ; 8.02 ηg ml-1) and 3.62 ηg ml-1 for the CQ-resistant (CQ; 65.35 ηg ml-1). The dual drug‟s activity was also superior to that of 1:1 mixture of artesunate and 4,7-dichloroquinoline, its precursors. The in vivo antiplasmodial activity of the dual drug was studied against CQ-sensitive P. berghei ANKA-infected mice. The drug produced significant dose-dependent activity against the parasite in the suppressive studies, with an ED50 and ED90 of 5.5 and 13.5 mg kg-1, respectively. The drug was also active against lumefantrine (LU)-sensitive and -resistant parasites with a suppressive effect of 98% (LU; 84%) and 67% (LU; 0%), respectively. The fact that the drug was equally active (p>0.05) against both CQS and CQR strains of P.
falciparum, and in vivo against both LUS and LUR strains of P. berghei point to its
potential as a suitable replacement of ALU (Coartem®) in case of pervasive resistance against the quinoline partner (LU) in ACT. The Dual drug showed a good safety profile, since at 10 000 ηg ml-1 (the highest concentration used) the drug showed low inhibition of Hep2 cell proliferation (29.3%), and IC50 values >10 000 ηg ml-1
CHAPTER ONE
INTRODUCTION
1.1 Background of the Study
Chemotherapy has been the mainstay of malaria control strategy especially in the absence of effective malaria vaccine. However, Plasmodium falciparum, the parasite that causes over 90% of all global malaria cases is increasingly becoming resistant to classical antimalarials, necessitating an urgent need for search and development of new antimalarial agents, preferably with novel mode of action (Ridley, 2002; Rosenthal, 2003). In the past two decades, only a few compounds belonging to a new class of antimalarial drugs, including amino alcohols (mefloquine (1), halofantrine (2), lumefantrine (3), sesquiterpene trioxanes (artemisinin derivatives), and naphthoquinones (atovaquone, 4) were developed for clinical usage (Basco et al., 2001).
N CF3
HO N H CF3 N OH F3C
Cl Cl
C4H9 C4H9
1 2
Cl
Cl N
HO
C4H9 C4H9
3 Cl O O OH 4
Organization (WHO) gold standard against P. falciparum malaria, in which the regimen uses a double or triple combination therapy geared towards delay of resistance, or circumventing it altogether (Capela et al., 2009; Arau´jo et al., 2009). However, recent study indicates that drug-resistant malaria parasites have spread to critical border regions of Southeast Asia. The study confirms that resistance to the world‟s most effective antimalarial drug, artemisinin, is now established in Western Cambodia, Thailand, Vietnam, Eastern Myanmar and Northern Cambodia (Ashley et al., 2014).
Various approaches to antimalarial drug discovery currently being deployed include optimization of therapy with available drugs including combination therapy, developing analogs of the existing drugs, evaluation of potent agents from natural products especially plants, use of compounds originally developed against other diseases, evaluation of drug-resistance reversers (chemosensitizers) as well as new chemotherapeutic targets (Rosenthal, 2003; Kouznetsov and Gomez-Barrio, 2009).
1.2 Problem statement
Plasmodium falciparum, the parasite that causes over 90% of all global malaria cases is
increasingly becoming resistant to classical antimalarials in clinical use, which poses a very significant threat in the fight against malaria. This dire situation is being aggravated by recent reports from South East Asia confirming that resistance to the world‟s most effective antimalarial drug, artemisinin, is now established in Western Cambodia, Thailand, Vietnam, Eastern Myanmar and Northern Cambodia (Ashley et al., 2014). Pervasive failure rates associated with ACT could greatly disrupt the current malaria elimination and eradication efforts, and again foster an increase in malaria cases and deaths (Dondorp et al., 2009). Resistance to antimalarial drugs arises when spontaneously occurring mutants with gene mutations or amplifications which confer reduced drug susceptibility are selected, and are then transmitted. Although a vaccine would be the best bet in the fight against the disease, efforts to develop an effective malaria vaccine are yet to be successful and thus chemotherapy may remain the mainstay of malaria control strategy for a long time. Thus, search for novel malaria therapies is not only urgent but also should be based on a paradigm of novel compounds and/or molecular targets (Ridley, 2002; Rosenthal, 2003).
1.3 Justification of the study
combination protect each other against resistance (Arau´jo et al., 2009). For this reason, use of combination therapy is the norm in management of infectious diseases.
There is need therefore to validate the concept of dual or hybrid drug, where two molecules with individual intrinsic activity are linked into a single agent, thus packaging dual-activity into a single hybrid molecule. It is envisaged that building on pharmacophores of existing drugs already indicated for human application to develop hybrid drugs may not only lower drug development period and cost but will also reduce drug development attrition rates while achieving the ultimate objective of either delaying or circumventing development of resistance altogether.
1.4 Hypothesis
The synthesized novel hybrid antimalarial drug is effective against both drug-sensitive and-resistant malaria parasites in culture and in a mouse model, indirectly supporting the dual drug‟s bioavailability.
1.5 Objectives
1.5.1 General objective
The overall objective of this study is to synthesize a novel antimalarial dual-drug that is effective against drug-resistant malaria, and that is affordable.
1.5.2 Specific objectives
i. To synthesize a dual drug comprising the quinoline and trioxane pharmacophoric scaffolds.
CHAPTER TWO
LITERATURE REVIEW
2.1 Historical background of malaria
Malaria is the most widespread parasitic disease caused by protozoa parasite of the genus
Plasmodium, with malaria endemic regions encompassing approximately 40% of the global
human population (Ridley, 2002; Rosenthal, 2003). Traditionally, four Plasmodia species cause human malaria, P. falciparum, P. vivax, P. ovale and P. malariae (Basco et al., 2001), although the primate parasite P. knowlesi was recently established as the fifth causative agent of human malaria (Olliaro and Wells, 2009). P. falciparum is the most prevalent and virulent of these parasites, responsible for about 90% of all global malaria-associated deaths each year (Enato and Okhamafe, 2004).
towards reducing malaria morbidity and mortality (Gregson and Plowe, 2005). There have been concerted efforts to develop an effective malaria vaccine since mid-1980s.
RTS, S is the first of the current generation of malaria vaccines which was identified as a potential candidate for further development following encouraging results in an experimental challenge study (Stoute et al., 1997). In subsequent Phase II clinical trials in children, the vaccine was found to have a promising safety profile and provided protection against clinical episodes of malaria in the range of 30- 60%. It could also be administered together with the package of vaccinations routinely given to African children (Bojang et al., 2001). Phase III clinical trials initiated in 2009 through a unique public-private partnership of Glasko SmithKline (GSKBio), the PATH (Professional Association of Therapeutic Horsemanship), Malaria Vaccine Initiative (MVI) and African and other research organizations with funding support from the Bill and Melinda Gates Foundation.
2.2 Disease transmission
Two hosts are involved in the life cycle of malaria parasite; mosquito and human host (Cowman et al., 2012). A malaria-infected female Anopheles mosquito injects sporozoites into the human host during a blood meal (Sturm et al., 2006; Prudencio et al., 2011). Sporozoites infect liver cells and mature into schizonts, which rupture and release merozoites that infect red blood cell. The parasites then undergo asexual multiplication in the erythrocytes (Cowman et al., 2012). Some parasites differentiate into sexual erythrocytic stages (gametocytes). An Anopheles mosquito ingests the male and female gametocytes during a blood meal, whose union generates zygotes which develop in to motile and elongated ookinetes that invade the mid gut wall of the mosquito (Aly et al., 2009). Here, they develop into oocysts, which grow, rupture, and release sporozoites that make their way to the mosquito's salivary glands. The malaria life cycle continues when these sporozoites are injected into a new human host (Newton and White, 1999).
2.3 Malaria chemotherapy
The available antimalarials belong to three broad classes namely, the arylamino alcohol
compounds that include mefloquine (1), halofantrine (2), lumefantrine (3) chloroquine (CQ,
5), amodiaquine (6), and primaquine (7); the antifolates that include pyrimethamine (8), proguanil (9) and the artemisinins which include artemisinin (10) and its semisynthetic derivatives dihydroartemisinin (11), artesunate (12), artemether (13) and arteether (14) (Egan and Keschola, 2007; Parija and Praharaj, 2011). Of these, mefloquine (1), CQ (5), and primaquine (7) have been the most widely used drugs in the fight against malaria.
N
HN NEt2
Me
Cl
5
N Cl
HN NEt2
OH 6 N HN NH2 O 7 N N NH2 Cl
C2H5 H2N
8
NH N H2N
NH2 Me Me Cl 9 O O R Me Me Me O O
The increasing trend of resistance to these drugs in the past few decades has, however made them redundant in many parts of the world, which are endemic for P. falciparum infection
10, R =O
11, R = OH
12, R = αOCO(CH2)2CO2H 13, R = βOCH3
(Petersen et al., 2011). Furthermore, most of antimalarials in clinical application are either chemically related and/or share the same putative target, thus may possess similar toxicity and share cross-resistance (Foley and Tilley, 1998; Bloland, 2001).
To prolong the lifespan of artemisinin drugs by reducing the emergence of drug-resistant parasites, artemisinin derivatives are administered in combination with at least one other antimalarial compound (Noedl et al., 2008). The current trend is to co-formulate two or more agents into a single tablet, termed as multi-component drug as opposed to the traditional cocktail therapy, so as to improve patient compliance (Morphy and Rankovic, 2005).
World Health Organization (WHO) currently recommends use of semi-synthetic derivatives of artemisinin (10) (isolated from Chinese herb Artemisia annua).This include dihydroartemisinin (11), artesunate (12) and artemether (13) in combination with longer acting drugs as the first-line therapies against drug-resistant falciparum malaria. The rationale for this combination is that the artemisinin derivative rapidly clears 95% of the parasites and the remaining 5% are cleared by the longer half-life partner drug (White, 1999). These combinations are termed as artemisinin-based combination therapy (ACT), and include artemether-lumefantrine (Coartem®), artesunate-amodiaquine, artesunate-mefloquine, artesunate-sulfadoxine pyrimethamine and dihydroartemisinin-piperaquine (Eastman and Fidock, 2009).
contained, it is possible that resistance will spread into malaria endemic regions of Africa and could derail the global drive to control and eventually eliminate malaria altogether. Thus the urgency for search for novel antimalarial drugs and targets can never be gainsaid.
Quinine from Peruvian Cinchona trees provided the lead for the discovery and development of synthetic aminoquinolines, the most notable being chloroquine (5) (Wang et al., 2007; Cosle‟dan et al., 2008). Likewise, the discovery of artemisinin from the Chinese herb A.
annua has served as a template for development of semi-synthetic artemisinins including
artesunate and artemether, which are being used extensively in ACT against drug-resistant malaria (Maude et al., 2010). The commercial availability of artemisinin (and hence its semi-synthetic derivatives) is limited by the fact that its only source is from A. annua since to date, there is no fully synthetic peroxidic antimalarial drug that has been made available for clinical application, which is unfortunate because of limitations associated with artemisinin semi-synthetics. The limitations include chemical (availability, purity and cost), bio-pharmaceutical (poor bioavailability and limiting pharmacokinetics), and treatment (non-compliance with repeated regimens and recrudescence) issues that limit their therapeutic potential (Vennerstrom et al., 2004; Perry et al., 2006). As a result, extensive research into synthetic endoperoxide antimalarials drugs has been undertaken in the last 15 years to produce molecules that are structurally simpler and synthetically accessible with a projected low cost of goods (Sabbani et al., 2008).
choice of the linker can allow the intact hybrid to dissociate into its individual components or to remain fixed when metabolically resistant linker units are chosen. The first case is particularly advantageous if the individual components have different sites of action within the cell while the latter is desirable with respect to overcoming resistance (Walsh and Bell, 2009).
Hybrid molecules can be classified as:
a) Conjugates; in which a distinct linker group that is not found in either of the
individual drugs separates the molecular frameworks, which contain the pharmacophores for each target. Most conjugates contain a metabolically stable linker.
b) Cleavage conjugates have a linker designed to be metabolized to release the two
drugs that interact independently with each target.
c) Fused hybrid molecules have size of the linker decreased such that the framework of
the pharmacophores is essentially touching.
d) Merged hybrids have their frameworks merged by taking advantage of commonalities
in the structures of the starting compounds, which give rise to smaller and simpler molecules (Morphy and Rankovic, 2005).
Artemisinin and its derivatives have a chemical structure significantly different from that of quinoline-based drugs. The attention of different groups regarding the mechanism of action of these potent antimalarials devoid of significant clinical resistance up to date has been drawn (Meshnick et al., 1996; Robert et al., 2005). Artemisinin, with its 1,2,4-trioxane as active motif, has served as a source of inspiration for the design of synthetic peroxide-containing drugs (Vennerstrom et al., 2004; Posner et al., 2008). The quinoline nucleus has been a chemical reference of highly active antimalarial drugs for many decades and several effective drugs containing this entity such as mefloquine (1), CQ (5), amodiaquine (6) and primaquine (7) have been developed.
The underlying mechanism behind the therapeutic effectiveness of artemisinin based hybrid molecules is that the artemisinin derivative is active on the young erythrocytic stages of P.
falciparum and the CQ derivative is able to inhibit the polymerization of β-haematin
(Meunier, 2008).
antimalarial drugs against both asexual and sexual malarial stages, and their potency is independent of CQ-sensitivity of the target parasite (Benoit-Vical et al., 2007).
In a recent report it has been argued that there is need to develop novel antimalarial drugs that possess pleiotropic action (multiple targets/modes of action like. quinolines and artemisinins) as opposed to single target drugs like pyrimethamine and sulfadoxine, since historical evidence on development and evolution of resistance shows that the former always have longer useful therapeutic lives (Muregi, 2010). In addition, recent reports have suggested that hybrid drugs, pro-drugs and double pro-drugs may serve as important strategies that might revolutionize malaria chemotherapy in the future (Muregi and Ishih, 2010; Muregi et al.,
2011). Therefore, it is proposed that a dual-drug containing a trioxane and quinoline pharmacophores is an attractive strategy since the two are pleiotropic, and may aid in delaying development of resistance due to enhanced dual/multiple functionality.
CHAPTER THREE
MATERIALS AND METHODS
3.1 Chemicals, glassware and study site
The test drugs, artesunate, chloroquine, 4,7-dichloroquinoline and all other chemicals and reagents were purchased from Sigma Chemical Co., Dorset, United Kingdom. The glassware apparatus used were washed, rinsed with distilled water, and dried in an oven at 110oC. They were allowed to cool and then rinsed with absolute ethanol.
The study was carried out at the Centre for Traditional Medicine and drug Research (CTMDR), Kenya Medical Research Institute (KEMRI), Nairobi, Kenya.
3.2 Synthetic design of quinoline-trioxane hybrid molecule
The synthetic design is as illustrated in figure 3.1. In general, it involved introduction of a linker to 4,7-dichloroquinoline and subsequent coupling of the two molecules (Chollet et al., 2009). N N H Cl N H O O O O H3C
CH3 O O O 17 Artes unate (1.2 eq.), CDI or EDC (1.5 eq.), CH2
Cl2,
HOBt (eq),
DIPE A (1
.0 eq ),
0oC to ro
om te mpera ture CH3 N Cl Cl
Ethylene diamine (4.5 equiv) Neat
Heated at 80oC for 1 hr,
then at 110oC for 4-6 hrs N
N
Cl
H NH2
15 16
3.2.1 Introduction of a linker
To 4,7-dichloroquinoline 15 (1 mmol, 0.198 g) was added ethylene diamine (4.5 equiv, 0.3 ml) (Figure 3.1a) in a round bottomed flask. The flask was then fitted with a condenser and the suspension was heated at 80 oC for 1 h before elevating the temperatures to 110oC for 4-6 h with continued stirring with a magnetic stirrer to drive the reaction to completion. The reaction mixture was cooled to room temperature and diluted with dichloromethane (20 ml). The organic layer was successively washed with 5% NaOH (30 ml), water and finally with brine. The organic layer was dried over anhydrous Na2SO4 and the solvent removed under reduced pressure in a rotary evaporator to afford the intermediate 16 (Figure 3.1a) (Antinarelli et al., 2012). The resulting powder was used for the next step without further purification.
N Cl
Cl
Ethylene diamine (4.5 equiv) Neat
Heated at 80oC for 1 hr,
then at 110oC for 4-6 hrs N
N
Cl
H NH2
15 16
Scheme 3.2:Introduction of a linker
3.2.2 Coupling procedure
overnight (about 15-16 h). Upon completion of the reaction, the reaction mixture was quenched with saturated NaHCO3 (20 ml). The organic phase was separated and the aqueous phase back-extracted with CH2Cl2 (20 ml × 3). The combined organic layers were dried using anhydrous Na2SO4, filtered and the solvent removed in vacuo. Purification using silica gel column pretreated with ammonia yielded the desired hybrid drug 17. The reaction was optimized by using other solvents like acetone and THF to dissolve the artesunate instead of CH2Cl2, and DMAP (additive), instead of HOBt and other coupling agents like DCC and CDI instead of EDC.
N N
Cl
H NH2
16 N N H Cl N H O O O O H3C
CH3 O
O O
17
Artesunate (1.2 eq.) EDC (1.5 eq.),CH2Cl2, HOBt(eq), DIPEA (1.0 eq), 0oC to room temperature
CH3
Scheme 3.3:Coupling reaction
3.3 General instrumentation
impact mass measurement was recorded on a GC/mass spectrometer machine (VG–12–250). Thin layer chromatography (TLC) was performed using pre-coated silica gel 60 F-254 glass plates (Merck). The solvent system used was ethyl acetate:methanol 3:1 with a drop of ammonia. Silica gel 5-40 μm mesh (Merck, Germany) was used for column chromatography with a solvent system of ethyl acetate:methanol 2:1 and a few drops of ammonia. The solvents were redistilled to ensure purity.
3.4 Bioassays of the hybrid drug
3.4.1 In vitro assays
3.4.1.1 Drug solutions
Stock solution of the hybrid drug was prepared with sterile water (deionized and autoclaved) and filter-sterilized through 0.22 μm filters under laminar hood. Stock solutions of reference drugs CQ, artesunate as well as 4,7-dichloroquinoline was similarly prepared in sterile water. Dissolution of the drugs which was insoluble in water was enhanced by first dissolving them in dimethylsulfoxide (solvent concentration <0.02%) (Elueze et al., 1996). The test samples were prepared as a 2 mg ml–1 stock solution and sonicated to enhance solubility then stored at 4 °C. Further dilutions were prepared on the day of biological assays. All the drug solutions were stored at 4°C for later use.
3.4.1.2 Plasmodium falciparum cultures
N-2-hydroxyethylpiperazine-N-2-ethanesulfonic acid (HEPES) and 25 mM NaHCO3. Human type O+ erythrocytes (<28 days old) served as host cells and the cultures were incubated at 37 °C in an atmosphere of 3% CO2, 5% O2 and 92% N2 obtained from BOC Nairobi, Kenya.
3.4.1.3 Evaluation of antiplasmodial activity of the hybrid drug
Aliquots (25 l) of the culture medium were added to all the wells of a 96-well flat-bottom micro-culture plate. The hybrid drug solution in volumes of 25 l was added in triplicate, to the first wells and a Titertek motorized hand diluter (Flow Laboratories, Uxbridge, United Kingdom) was used to make serial two-fold dilutions of each sample over an 11-fold concentration range. The highest concentration of 100 ηg ml–1was serially diluted 2-fold in complete medium to give eleven doubling concentrations with the lowest concentration being 0.0975 ηg ml–1. The same dilution technique was used for the reference drugs CQ and artesunate samples except 4,7-dichloroquinoline whose starting concentration was 1000 ηg ml–1.
The in vitro semi-automated micro-dilution assay technique that measures the ability of the
was measured by liquid scintillation.
The concentration causing 50% inhibition of radioisotope incorporation (IC50) was determined by interpolation after logarithmic transformation of both concentration and cpm values using the formula;
IC50 = antilog (log X1 + [(logY50-log Y1) (logX2-logX1)]/(logY2-logY1)]…….Equation 3.1, where Y50 is the mid-point, X1, X2 are lower and upper concentrations respectively, and Y1, Y2 are the corresponding cpm values for the data points above and below the cpm mid-points (Sixsmith et al., 1984).
3.4.1.4 Combination studies of the individual precursors of the hybrid drug
To compare the antiplasmodial activity of the hybrid drug (100 ηg ml–1) with that of the combination of its precursors, wells of the combination of the latter were also included in the test in the ratio of 1:1 (mixture of 50 ηg ml–1 of artesunate and 50 ηg ml–1 of 4,7-dichloroquinoline). Subsequent serial dilutions and semi-automated micro-dilution assays as outlined in section 3.3.1.3 above were done. Since it is impossible to determine a single IC50 of two drugs in combination, the cpm values for the wells of hybrid drug and for the drugs in combination at a given concentration were used to determine percentage in vitro inhibition of parasite proliferation, relative to drug-free parasitized erythrocyte wells.
3.4.2. In vivo assays
3.4.2.1 Parasite, host and ethics
Plasmodium berghei-ANKA isolates (wild type and lumefantrine-resistant) were used for this
The day of infection was denoted as day zero post infection (pi) and all experiments were done using this revived parasite. At day 5 pi, the donor mouse parasitaemia was assessed microscopically (Olympus BX50F4, Olympus Optical Co., LTD., Japan) at 1000× magnification by examining Giemsa stained thin tail-vein blood smears. The mouse was later sedated, bled via cardiac puncture and blood collected in heparinized tubes. The parasitaemia was adjusted downwards using phosphate saline glucose (PSG) buffer and each of the test Swiss albino mice 6 weeks old (weight 20±2 g) was inoculated ip with approximately 1x107 parasitized erythrocytes in volumes of 0.2 ml (Muregi et al., 2008). The inoculated mice were then randomized into group of 5, housed in cages and maintained in the animal facility on commercial rodent pellets and water ad libitum.
This work was fully approved by KEMRI-Animal Care and Use Committee (ACUC) approval number SSC 2592 (Appendix2) and all experiments conducted in accordance to the Institute‟s guide for the care and use of laboratory animals. All mice that were deemed to have completed their intended use were euthanized in the course of experiments, placed in biohazard disposable autoclave bags and autoclaved at 121ºC for 15 minutes before incineration to destroy all infectious agents and to avoid environmental contamination during incineration.
3.4.2.2 Drug vehicle
3.4.2.3 In vivo antiplasmodial evaluation of the hybrid drug
To assess the antiplasmodial effect of the hybrid drug in mice, the 4-day suppressive method of Peters et al., (1975) was adopted. Within 3 hours post inoculation of mice with the parasites (that is on day 0), treatment of the experimental groups was initiated by oral administration at a dose range of 50 mg kg-1 body weight (highest dose) to 1.25 mg kg-1 body weight (lowest dose) given once a day for 4 days, up to 3 days pi. Two positive control groups were treated with artesunate (at doses of 10, 5, 2.5, 1.25 and 0.625 mg kg-1 body weight) and 4,7-dichloroquinoline (at doses of 50 mg kg-1 body weight, 25, 12.5 and 6.25 mg kg-1 body weight). Untreated control groups that received the drug vehicle only were also included. On day 4 pi, parasitaemia of individual mouse was determined by microscopic examination of Giemsa stained thin blood smears prepared from mouse tail blood. Smears were done on day 7, 9, 11 and 15 pi to monitor parasitaemia. Microscopic counts of blood films from each mouse were processed using Microsoft® Excel (Microsoft Corp.). Percentage (%) chemosuppression (parasite reduction) of each dose was determined as described by Tona et al. (2001).
…...Equation 3.2,
where A is the mean parasitaemia in the negative control group and B is the parasitaemia in the test group. The ED50 and ED90 values (the concentration which inhibited 50% and 90% parasite growth, respectively) for each drug were estimated graphically by linear regression using version 5.5 of Statistica 2000.
3.4.3 In vivo combination studies for the hybrid drug precursors
an inoculum of 1x107 P. berghei ANKA parasites as detailed in subsection 3.4.2.3 were treated orally for 4 days with different combinations of the two drugs. The first combination comprised the ED50s of artesunate and 4,7-dichloroquinoline. The second combination had half (1.14 mg kg-1 of artesunate: 23.5 mg kg-1 of 4,7-dichloroquinoline) of the first respective ED50s while the third combination had a quarter (0.57 mg kg-1 of artesunate: 11.75 mg kg-1 of 4,7-dichloroquinoline) of the first combination. Percentage (%) chemosuppression of each combination was determined as outlined in subsection 3.4.2.3.
3.4.4 Studies of the hybrid drug against lumefantrine-resistant parasites
The hybrid molecule was also tested against lumefantrine (LU)-sensitive (LUS, wild type) and -resistant (LUR) parasites using the 4-day suppressive method of Peters et al. (1975) as detailed in subsection 3.4.2.3. LU-resistance was generated artificially through subjecting wild type P. berghei ANKA parasite to LU selective pressure for 73 passages (Kiboi et al., 2009), and the parasites were obtained from KEMRI. A dose of pre-determined ED90 (3.93 mg kg-1) for LU against wild type parasite was used to orally treat two groups of Swiss albino mice infected ip with wild type and LU-resistant P. berghei ANKA at an inoculum of 1x107 parasites. Similarly, the pre-determined ED90 (13.5 mg kg-1) for the hybrid molecule against wild type parasite was used to orally treat two groups of mice infected with the LUS and LUR parasites, and percentage (%) suppression determined as detailed in subsection 3.4.2.3.
3.4.5 Cytotoxicity assays
cells. The amount of generated formazan is directly proportional to the number of cells (Mosmann, 1983).
The human larynx carcinoma (Hep2) cells were used for this study. Cells were maintained in Eagles Minimum Essential Medium (MEM) containing 10% fetal bovine serum (FBS). A cell density of 20,000 cells per well in 100 µl were seeded on 96-well plates and incubated for 12 hours at 37 °C and 5% CO2 to attach to the surface. Samples of the test drug and controls were added to the cultured cells in rows H - B over a concentration range of 0.14 to 100 µg ml-1, whereas wells 1-8 row A served as untreated controls and wells 9-12 as blank (1% DMSO v/v). The plates were incubated for 48 hours at 37°C and 5% CO2, followed by addition of 10 µl MTT viability indicator reagent, and were incubated for additional 4 hours at the same conditions. All media was removed from the plates and 100 µl DMSO was added to dissolve the formazan crystals. The plates were read on a Multiskan EX Labsystems scanning multi-well spectrophotometer at 562 and 690 nm as reference. The results were recorded as optical density (OD) per well at each drug concentration. The data was transferred into the software Microsoft Excel 2007 and expressed as percentage of the untreated controls.
Percentage cytotoxicity (PC) as compared to the untreated controls was calculated using the following equation:
Percentage cytotoxicity = [(A – B)/A] × 100……Equation 3.3,
3.5 Data analysis
NMR spectra for both 13C and 1H was analyzed using characteristic chemical shifts in ppm while for IR characteristic peaks in wavenumbers generated were used. In mass spectra, the peaks for the molecular ion and major fragments were analyzed.
Data for in vitro drug assays were transferred into graphic programme (Microsoft Excel 2007) and results expressed as the drug concentration required for 50% inhibition of (G-3H) hypoxanthine incorporation into parasite nucleic acid using non-linear regression analysis of the dose-response curve.
For in vivo data, percentage parasitaemia was recorded in Excel (Microsoft) and expressed as
CHAPTER FOUR
RESULTS AND DISCUSSION
4.1 Structure elucidation of 7-chloro-4-(1, 2-diaminoethyl)quinoline (16)
Compound 16 was isolated as a yellow powder (0.186g, yield 84%, Rf 0.6 SiO2, 1:3, EtOAc: MeOH, mp 142-145oC). The IR spectra showed characteristic bands at Vmax 3361(-N-H), 1585(-C=C-) and 1326(-C=N-) (Appendix 3).
N
N
Cl
H
NH
216
2 2' 3 4 5 6 8 1' 8a 4aThe 1H-NMR spectrum of compound 16 showed five different protons in the aromatic region at δ = 6.40 (d, J = 5.4 Hz, 1H, H-3), δ = 7.39 (dd, J = 9.0 and 1.8Hz, 1H, H-6), δ = 7.75 ( d, J
= 9.2Hz, 1H, H-5), δ = 7.95 (d, J = 1.8Hz, 1H, H-8 and δ = 8.52 ( d, J = 5.6 Hz, 1H, H-2) ppm. The methylene protons showed broad multiplets at δ = 3.36 (m, 2H, H-1‟) and a triplet at δ = 3.14 (J = 4.8 Hz, 2H, H-2‟) ppm. The 1H-NMR data of compound 16 is summarized in table 4.1.
Table 4.1: 1H-NMR (200MHz) data of compound 16 in CDCl3
Position Chemical shift δ (ppm) Multiplicity J (Hz) Integral
1‟ 3.36 (3.43)* m - 2H
2‟ 3.13 (2.97)* t 4.8 (6.4)* 2H
2 8.52 (8.35)* d 5.6 (5.6)* 1H
3 6.40 (6.55)* d 5.4 (5.6)* 1H
5 7.75 (8.10)* d 9.2 (9.0)* 1H
6 7.35 (7.39)* dd 8.8, 2.1(9.0, 2.1)* 1H
8 7.95(7.76)* d 1.8 (2.1)* 1H
The proton assignments were further confirmed by COSY experiments of compound 16
(Figure 4.1).
N N
NH2
Cl H
H
H
H H
H H
H H
Figure 4.1: Correlations observed in H-H COSY of compound 16
The 1H-NMR data of compound 16 (Appendix 3A) compared closely with reported data of the 7-chloro-4-(1, 2-diaminoethyl)quinoline (N‟Da and Breytenbach, 2011).
The 13C-NMR spectrum (Appendix 3B) for compound 16 revealed 11 carbon signals as two methylene, five methine and four quaternary carbons. The two methylene signals at δ = 40.4 and 44.9 ppm were determined to be aliphatic whereas the signals for aromatic carbons ranged from δ = 99.7 to 152.3 ppm. The DEPT spectrum showed all protonated carbon atoms with those corresponding to CH2 group at δ = 44.9 and 40.4 ppm. The chemical shifts corresponding to CH were observed at δ = 99.7, 121.5, 125.5, 128.9 and 152.3 ppm whereas the quaternary carbons were observed at δ = 117.6, 135.1, 149.3 and 150.2 ppm. The 13
Table 4.2:13C-NMR (50MHz) data of compound 16 in CDCl3
Position Chemical shift δ (ppm)
Chemical shift of 7-chloro-4-(1, 2-diaminoethyl)quinoline (ppm))
DEPT
1‟ 44.9 46.3* CH2
2‟ 40.4 40.8* CH2
2 152.3 152.5* CH
3 99.7 99.7* CH
4 150.2 152.8* C
4a 117.6 118.8* C
5 128.9 127.6* CH
6 125.5 126.0* CH
7 135.1 136.3* C
8 121.5 124.4* CH
8a 149.3 149.7* C
*N‟Da and Breytenbach (2011).
Compound 16 was identified to be 7-chloro-4-(1,2-diaminoethyl)quinoline with molecular formula of C11H12N3Cl by electron spray ionization mass spectrum that showed molecular ion peaks at m/z 221.8 and 223.9 amu against a calculated value of 221.07. From the MS spectrum (Appendix 3C), the presence of one chlorine atom can be deduced by the presence of two peaks separated by two mass units that is 221.8 and 223.9 amu due to the two isotopes of chlorine ( 35Cl and 37Cl). The 1H-NMR and 13C-NMR spectral data given in table 4.1 and table 4.2 were in agreement with reported literature values for 7-chloro-4-(1,2-diaminoethyl)quinoline (Lombard et al., 2011).
4.2 Structure elucidation of N-(7-chloroquinolin-4-ylamino) ethylartesunate-19- carboxamide (17)
Table 4.3: Percentage (%) yield of compound 17 using various coupling reagents
Entry Coupling Reagent Solvent Additives (eq) Yield (%)
1 EDC THF - 11
2 EDC Acetone - Trace
3 EDC THF DMAP (0.2) 17
4 EDC CH2Cl2 - 30
5 EDC CH2Cl2 HOBT (0.2) 71
6 DCC CH2Cl2 - 8
7 DCC CH2Cl2 HOBT (0.2) 13
8 CDI CH2Cl2 - 14
9 CDI CH2Cl2 HOBT (0.2) 51
The IR spectra showed characteristic bands at Vmax 3361H), 1747 (-C=O), 1699 (-N-C=O), 1585 (-C=C-) and 1326 (-C=N-) (Appendix 4).
The 1H-NMR spectrum (Appendix 4A) of compound 17 revealed three methyl proton groups at δ = 1.34 (s, 3H, H-14) ppm; δ = 0.74 (d, J = 9.6 Hz, 3H, H-15) ppm and δ = 0.87 (d, J = 9.2 Hz, 3H, H-16) ppm. Five different peaks were observed in the aromatic region that is δ = 8.50 (d, J = 3.2 Hz, 1H, H-2‟) ppm, δ = 6.31 (d, J = 3.6 Hz, 1H, H-3‟) ppm, δ = 7.91 (d, J = 3.6 Hz, 1H, H-5‟) ppm, δ = 7.39 (dd, 1H, H-6‟) and δ = 7.89 (d, J = 12.0 Hz, 1H, H-8‟) ppm. The methylene and methine protons on the artemisinin rings (C-4, C-5, C-5a, C-6, C-7, C-8, C-8a and C-9) and the aliphatic protons (C-19 and C-20) appeared as broad multiplets from δ = 0.94-2.78 ppm.
N N H Cl N H O O O O H3C
The 1H-NMR chemical shifts are summarized in table 4.4.
Table 4.4:1H-NMR (400 MHz) chemical shifts of compound 17 in CDCl3
Position Chemical shift δ (ppm) Multiplicity J (HZ) Integral
2‟ 8.50 d 3.2 1H
3‟ 6.31 d 3.6 1H
3‟‟ 3.43-3.71 m - 2H
5‟ 7.91 d 3.6 1H
6‟ 7.39 dd 2.8 and 12 1H
8‟ 7.89 d 12 1H
10 4.76 d 12 1H
12 4.88 s - 1H
14 1.34 s - 3H
15 0.74 d 9.6 3H
16 0.87 d 9.2 3H
The 13C-NMR spectrum (Appendix 4B) revealed 30 carbon signals as three methyl, eight methylene, eleven methine and eight quaternary carbons as summarized in table 4.5. Four methylene were determined to be aliphatic and their chemical shifts were observed at δ = 28.2-45.4 ppm. Four other methylene carbon atoms were determined to be cyclic with chemical shifts ranging from δ = 20.3-36.4 ppm. The characteristic chemical shifts for aromatic carbon atoms were observed at δ = 98.5-151.9 ppm. Two signals in downfield at δ = 178.0 and 173.7 ppm were assigned to amide carbonyl carbon and ester carbonyl carbon respectively.
peaks separated by two mass units that is 588.24 and 590.24 amu (Appendix 4D) due to the two isotopes of chlorine (35Cl and 37Cl).
Table 4.5: 13C-NMR (100MHz) data of compound 17 in CDCl3
Position Chemical shift (ppm) Position Chemical shift (ppm)
2‟ 151.7 6 37.4
3‟ 98.5 7 34.4
3‟‟ 45.4 8 20.3
4‟ 117.3 8a 44.9
4‟‟ 42.8 9 31.5
5‟ 122.4 10 92.4
5‟a 52.5 12 91.1
6‟ 125.7 12a 80.4
7‟ 135.2 14 25.8
8‟ 150.3 15 11.8
8‟a 127.7 16 20.0
3 104.4 18 173.7
4 36.4 19 31.5
5 24.5 20 28.2
5a 51.5 21 178.0
The 1H-NMR and 13C-NMR spectral values for compound 17 compared to reported literature values for the artesunate part of compound 18, and for the quinoline part of compound 19 (Chollet et al., 2009; N‟Da and Breytenbach, 2011).
N O H O O O CH3 CH3 O
H3C O O
18 N H N N N NH2
H2N
H N N H O O CH3 CH3 O
H3C O O
19
N Cl
Compound 19: {2-[(7-chloroquinolin-4-yl)amino] ethyl}(2- {(10- dihydroartemisinin-10-yl)oxy}ethyl) amine (N‟Da and Breytenbach, 2011) was used for comparison purposes.
The structure is further supported by mass fragmentation patterns and rearrangements shown in Scheme 4.1.
N N H Cl N H O O O O H3C
CH3 O O O 17 CH3 O O CH3 CH3 CH3 HN H N O O O O O N Cl N N OH O O H H m/z 322 m/z 435
In this study, artesunate was coupled to an aminoquinoline moiety with the aim of validating the concept of covalent bitherapy. Covalent biotherapy is the chemical combination of two distinct pharmacophores into a single molecule, the so-called hybrid molecule (N‟Da and Breytenbach, 2011), which is an emerging strategy within medicinal chemistry and drug discovery. The hybrid molecule offers a simpler and more effective way of delivering two drugs, especially when differences like elimination times occur (Walsh and Bell, 2009).
There are numerous advantages of employing hybrid molecules over multi-component drugs in malaria therapy. Compared to the latter, hybrid drugs may be less expensive since in principle, the risks and costs involved may not be different from any other single entity and also there is a lower risk of drug-drug adverse interactions compared to multi-component drugs (Morphy and Rankovic, 2005). It is also expected that the hybrid drug would be metabolized by the liver enzymes to release both pharmacophores thus giving rise to dual action, possess increased activity and prolonged half-life (Arau´jo et al., 2009). However, it should be noted that to date no hybrid drug has reached clinical application stage.
latter which are lost to resistance within a short period of their therapeutic use (Muregi, 2010). Therefore, it is proposed that a dual-drug containing a trioxane and quinoline pharmacophores is an attractive strategy since the two are pleiotropic, and may aid in delaying development of resistance due to enhanced dual/multiple functionality.
In this study, the quinoline pharmacophore, 4,7-dichloroquinoline was covalently linked to artesunate via a diaminoalkyl linker. Variations in the length of the linker between the trioxane and the 4-aminoquinoline moiety have been shown to affect the activity of the hybrid molecule with lower activity if the alkyl chain is longer than two carbon atom units (Arau´jo et al., 2009), which informed the choice of a two carbon unit linker in this study. It should be noted that for artemisinins, the pharmacophore (1,2,4-trioxane group) which confers artemisinins with their unique activity, has not been synthesized to date. Therefore artesunate was used to provide the trioxane moiety which was successfully conjugated to the quinoline in a 2-step process (Figure 3.1) to afford the artesunate-quinoline hybrid. The ease of synthesis of the dual drug should also be considered an additional advantage since it would make it affordable and without perturbations to drug supply, if the drug is shown to be efficacious and safe in clinical studies.
4.3 Biological studies of the hybrid drug
4.3.1 In vitro studies
4.3.1.1 In vitro inhibition of Plasmodium growth by the hybrid drug
Table 4.6: IC50 of dual-drug, artesunate, chloroquine (CQ), 4,7-dichloroquinoline (DQ) against CQ-sensitive (D6) and -resistant (W2) isolates
Drug IC50 (ηg ml-1) SD (D6, CQSa) IC50 (ηg ml-1) SD (W2, CQRb)
Dual-drug 6.89 0.35 3.62 0.32
Artesunate 6.67 0.07 4.04 0.19
CQ 8.02 0.23 65.35 1.07
4,7-DQ > 1000 > 1000
aCQS, chloroquine sensitive; bCQR, chloroquine resistant
It was established that the drug was active in vitro independently of the susceptibility of the
P. falciparum strains to CQ. The drug‟s activity (IC50 of 6.89 ηg ml-1) against CQS (D6) was
comparable to that of artesunate (IC50 of 6.67 ηg ml-1), (p = 0.46) which is the most potent drug in the artemisinin family. The hybrid drug is significantly (p = 0.004) more potent on CQR parasites compared with CQS parasites (IC50, 3.62 ηg ml-1 vs 6.89 ηg ml-1) and about 18 times more active than CQ against CQR parasites (Table 4.6). This finding is very significant in that new drugs are urgently needed in areas where malaria is endemic and where CQ resistance is widespread (White, et al., 1999; Mutabingwa, et al., 2005).
4.3.1.2 Potency of the hybrid drug and its precursors
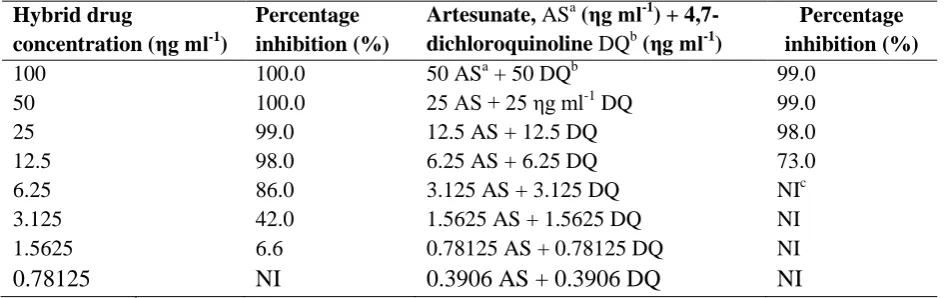
To validate the concept of dual molecules, the precursors, 4,7-dichloroquinoline and artesunate were tested in combination in the same wells. The inhibition (%) of parasite growth by the hybrid drug and precursors (artesunate and 4,7-dichloroquinoline) in combination are presented in table 4.7.
Table 4.7: In vitro percentage inhibition of parasite growth by the hybrid drug and precursors (artesunate and 4,7-dichloroquinoline) in combination
Hybrid drug
concentration (ηg ml-1)
Percentage inhibition (%)
Artesunate, ASa (ηg ml-1) + 4,7-dichloroquinoline DQb (ηg ml-1)
Percentage inhibition (%)
100 100.0 50 ASa + 50 DQb 99.0
50 100.0 25 AS + 25 ηg ml-1 DQ 99.0
25 99.0 12.5 AS + 12.5 DQ 98.0
12.5 98.0 6.25 AS + 6.25 DQ 73.0
6.25 86.0 3.125 AS + 3.125 DQ NIc
3.125 42.0 1.5625 AS + 1.5625 DQ NI
1.5625 6.6 0.78125 AS + 0.78125 DQ NI
0.78125 NI 0.3906 AS + 0.3906 DQ NI
a
AS, artesunate; bDQ, 4,7-dichloroquinoline; cNI, no inhibition
The percentage inhibition of parasite growth by the hybrid drug and precursors (Table 4.7) demonstrated that the hybrid drug is more potent than a combination of its precursors at comparable concentrations. For higher concentration (100-12.5 ηg ml-1), the inhibition for the dual drug is comparable to that of the combination of its precursors (almost 100%).However, at lower concentrations of the dual drug (6.25 ηg ml-1, % supression 86%; 3.125 ηg ml-1, % suppression 42%; 1.5625 ηg ml-1; % suppression 6.6%), there was significant activity (p<0.05) while the combination of its precursors had no activity at all, suggesting that the linker between the two molecules improves the dual drug‟s antiplasmodial activity, findings that are consistent with those of Benoit-Vical and colleagues (2007).
in vitro antiplasmodial activities (IC50 values) ranging from 4–32 nM, activities that were independent of the CQ sensitivities of the P. falciparum isolates tested. These activities were comparable to those of artesunate, and to test the concept of dual activity of the molecules, the quinoline and trioxane precursors were tested individually and in combination, and their activities compared with that of the conjugates (Benoit-Vical et al., 2007). Irrespective of the
P. falciparum strain used, the trioxane entity alone had IC50 values ranging from 200–600
nM, while the IC50 values of the quinoline motif alone ranged from 120 nM to 2 µM. In combination in the same well however, both entities had IC50 values ranging from 40-180 nM. The fact that the trioxaquines IC50 values were low (4-32 nM) implies that the link between both pharmacophores of the trioxaquines is essential for their activity (Benoit-Vical
et al., 2007). Walsh et al. (2007) demonstrated that a hybrid, artemisinin covalently linked to
quinine via an ester linkage had superior activity to that of artemisinin alone, quinine alone, or a 1:1 mixture of artemisinin and quinine, results that are consistent with our findings. This further confirms that the linker between both pharmacophores of the trioxaquine is essential for their activity.
N`DA and Breytenbach (2011) synthesized artemisinin-quinoline hybrids and tested them in
vitro against CQS (D10) and CQR (Dd2) P. falciparum strains. Their IC50 range (0.021-0.034
artesunate, the drug that provided the trioxane moiety of the dual drug. It should however be noted that the essence of drug combination or covalent bitherapy is not necessarily to enhance drug potency but to delay or circumvent resistance by using two or more components with different mechanisms of action or molecular targets.
4.3.2 In vivo studies
4.3.2.1 In vivo inhibition of Plasmodium growth by the hybrid drug
In vivo, the antiplasmodial activity of the dual drug administered orally at different doses was
determined using Peters 4-day suppressive test (Peters, 1975) using mice infected with the erythrocytic P. berghei parasite (Table 4.8).
Table 4.8: In vivo antiplasmodial activities* of the hybrid drug, artesunate and 4,7-dichloroquine against P. berghei in mice using the suppressive test
Drug/
Dosage (mg kg-1 day-1)†
Day 4 post-infection
Percentage suppression
Day 7 post-infection
Percentage suppression Hybrid drug 50 25 12.5 10 6.25 5 2.5 1.25 100.0 95.4 93.7 68.9 63.6 52.6 40.1 19.9 70.5 42.5 30.6 20.4 18.1 17.8 NS NS Artesunate 10.0 5.0 2.5 1.25 0.625 95.8 89.3 66.3 27.2 22.8 43.8 30.7 NS NS NS 4,7-dichloroquinoline 50 25 12.5 6.25 52.4 27.2 1.7 NS 53.0 17.5 18.0 18.3
Chloroquine (CQ) 100 100
The highest dose of 50 mg/kg exhibited suppressive effect of 100%, activity comparable to that of conventional antimalarials CQ and artesunate, which had 100% and 95.8% parasite growth suppression, respectively. The suppressive effect was dose dependent on day 4 pi and day 7 pi as 25, 12.5,10, and the lowest dose of 1.25 mg kg-1 produced a suppression of 95, 94, 69 and 20% respectively, while corresponding suppression by the same doses were 43, 31, 18% and no suppression with the lowest dose of 1.25 mg kg-1 on day 7 pi (Figure 4.1 and Table 4.8).
Figure 4.2: Dose-activity relationship of the hybrid on day 4 and 7 post infection (pi)
which is not surprising considering the very short half-life for artemisinins. However the suppressive effect of 4,7-dichloroquinoline is more or less maintained on day 4 and 7 pi, with values of 52 and 27% on day 4 pi (for 50 mg kg-1 and 25 mg kg-1 respectively), 53 and 18% on day 7 pi (Table 4.8). It can therefore be ascertained that the quinoline moiety (4,7-dichloroquinoline) confers longer half-life to the dual drug while the trioxane moiety (provided by artesunate) imparts rapid parasite clearance to the dual drug. Thus, the key overarching goal of developing a hybrid drug aligns with the primary reason of combining two drugs (co-formulation) in malaria chemotherapy, which is essentially to delay development of drug resistance by using two or more different drugs with different molecular targets. In such a scenario, the quick-acting drugs (artemisinins) rapidly reduce the parasite load while the longer acting drugs (quinolines) clear the lingering parasites (White et al., 1999). The ED50 and ED90 values at day 4 pi were determined as 5.5 and 13.5 mg kg-1 respectively as shown in figure 4.3.
Figure 4.3: Effective dose-50 (ED50) and ED90 values for the hybrid drug.
*P. berghei infected mice were orally treated once daily with 10, 5, 2.5 and 1.25 mg kg-1 bwt. of the dual drug between day 0 and 3 post infection and percentage suppression used to determine ED50 and ED90.
Artesunate was highly active with a suppression of 89.3% for the highest dosage used (5mg kg-1day-1) and 22.8% for the lowest dosage (0.625 mg kg-1 day-1). The ED50 for artesunate was determined as 2.27 mg kg-1 day-1. However, on day 7 the suppression for 5 mg kg-1 day-1 was only 30.7% while the rest of the dosages had no suppression. This is expected considering the very short plasma elimination time for artesunate.
With 4,7-dichloroquinoline, even the highest concentration (50 mg kg-1 day-1) had only about 50% suppression while the lowest (6.25 mg kg-1 day-1) had no suppression. The ED50 was determined as 47 mg kg-1 day-1. On day 7 pi, suppression is maintained with the highest dose (50 mg/kg/day) suppressing 53% and the lowest dose (6.25 mg kg-1 day-1) suppressing 18.3%. This is significant in that 4,7-dichloroquinoline appears to confer longer half-life to the dual drug, since a dose of 6.25 mg kg-1 (assumed to have 3.125 mg of artesunate and 3.125 mg of 4,7-dichloroquinoline, assuming 1:1 ratio) dual drug had a suppressive effect of 18% while a comparable dose of artesunate (2.5 mg kg-1) had no suppression on day 7 pi.
4.3.2.2 In vivo potency of the hybrid drug and its precursors
Table 4.9: In vivo antiplasmodial activity of artesunate (AS) and 4,7-dichloroquinoline (DQ) drug combinations on day 4 and 7 pi against P. berghei ANKA determined by the suppressive test in mice
Dosage (mg kg-1 day-1)*
Day 4 post-infection Day 7 post-infection
Mean Parasitaemia (±SD)
Percentage suppression
Mean Parasitaemia (±SD)
Percentage suppression
AS:DQ Combination
ED50:ED50 4.58 0.46 57.7 17.49 1.04 44.7
1/2ED50: 1/2ED50 8.20 0.93 21.6 25.36. 3.91 19.8 1/4ED50: 1/4ED50 9.82 1.27 8.9 36.91 1.54 NS Untreated control 10.78 0.84 - 31.62 0.53. -
*The drug combination was administered orally, once daily for 4 consecutive days; NS, no suppression A drug dose containing a combination of the individual precursors ED50S (ED50 of artesunate, 2.27 mg and ED50 of 4,7-dichloroquinoline, 47 mg) had a comparable inhibition of 58% (Table 4.9). However a dose of 2.5 mg/kg/day (assumed to contain 1.25 mg of artesunate and 1.25 mg of 4,7-dichloroquinoline in a 1:1 linkage ratio) had 40% inhibition, almost twice that of the combination of ½ED50S [½ ED50 of artesunate (1.14 mg kg-1) and ½ ED50 of 4,7-dichloroquinoline (23.5 mg kg-1)] which had 22% inhibition (Tables 4.8 and 4.9). The same trend is repeated with a dose of 1.25 mg kg-1 day-1 (assumed to contain 0.625 mg of artesunate and 0.625 mg of 4,7-dichloroquinoline in 1:1 linkage ratio) having 20% inhibition on day 4 pi compared with a combination of ¼ ED50`S (¼ ED50 of artesunate, 0.57 mg and ¼ ED50 of 4,7-dichloroquinoline, 11.75 mg) which had 9% inhibition. This confirms the earlier proposition that the linker in trioxaquines improves their antiplasmodial activity.
4.3.2.3 Potency of the hybrid drug against lumefantrine-resistant P. berghei in a mouse model