ABSTRACT
Cells must maintain genomic integrity despite constant exposure to sources of DNA
damage. The most genotoxic form of damage is the double-strand break and its improper
repair causes genomic instability and cancer predisposition. An undamaged homologous
sequence can serve as the template for repair of a double-strand break, and increased
mobility may be required to facilitate an efficient homology search. Chromatin exhibits
increased mobility upon DNA damage, but the physical mechanism remains unknown.
To explore this mechanism, we employ in vivo single-particle tracking of tagged loci in
haploid yeast. We find that cytoskeletal actin contributes to increased chromatin and
nuclear dynamics upon damage, reflecting its role in meiosis to promote homolog pairing
by nuclear mixing. This presents a new model for increasing chromatin mobility to
facilitate homology search in the DNA damage response.
INTRODUCTION
DNA is continually exposed to sources of damage including radiation,
carcinogens and replication errors. The double-strand break (DSB) is the most genotoxic
form of DNA damage (Agarwal, Tafel, & Kanaar, 2006; Wyman & Kanaar, 2006), and
its improper repair could lead to genomic instability, cancer predisposition or cell death
(Rassool, 2003; Thorslund & West, 2007). Therefore, the cell must have efficient and
accurate mechanisms for resolving DSBs.
Two mechanisms have been identified for repair of DSBs: nonhomologous end
joining (NHEJ) and homologous recombination (HR). Mammals rely largely on NHEJ,
key role in HR. When DNA becomes damaged, an undamaged homologous sequence can
serve as the template for repair, and, through the process of HR, the genetic information
lost by a DSB is restored. The undamaged sister chromatid is the preferred template for
repair of a DSB, but is not always readily available, and increased DSB mobility may be
required to facilitate an efficient homology search (Gehlen, Gasser, & Dion, 2011;
Savage, 1996). Recent studies have shown that chromatin exhibits such an increase in
mobility upon DNA damage (Dion, Kalck, Horigome, Towbin, & Gasser, 2012;
Miné-Hattab & Rothstein, 2012), but the mechanism remains unclear.
Homolog pairing also plays a key role in meiosis to ensure high fidelity of
chromosome segregation that haploidizes the genome prior to gametogenesis (Lee,
Conrad, & Dresser, 2012). How chromosomes become paired during meiosis is largely
unknown, but active nuclear stirring by the cytoskeleton has been suggested to play a role
in bringing homologous regions into proximity (Maguire, 1974). In S. pombe, telomeres
move along microtubules via dynein motors and become clustered at the spindle pole
body (SPB), forming the bouquet arrangement in preparation for meiosis (Chikashige et
al., 1994). Similar rapid movements of chromosomes have been identified in the budding
yeast S. cerevisiae, but appear to be promoted by actin rather than by microtubules
(Koszul, Kim, Prentiss, Kleckner, & Kameoka, 2008; Scherthan et al., 2007;
Trelles-Sticken, Adelfalk, Loidl, & Scherthan, 2005).
We investigated whether the actin-driven nuclear stirring mechanism implicated
in S. cerevisiae during meiosis to pair homologs is used in the DNA damage response (DDR) to facilitate homology search. We find nuclear dynamics depend on actin but not
suggest a role for actin in maintaining dynamics of the nucleus. It remains to be seen if
actin drives the increased nuclear motion observed upon DSB.
RESULTS
Chromatin’s response to DNA damage is global
We investigated how chromatin dynamics are altered during interphase in
response to two different DNA damage conditions: (1) multiple DSBs induced randomly
across entire chromosomes and (2) a single I-SceI-induced DSB at a known locus. To
measure, we use in vivo single-particle tracking of a GFP-labeled chromatin array
(lacO/lacI-GFP) at RAD16 relative to the unduplicated SPB (Spc29-RFP) over 10 min at
30 s intervals and measure subpixel localization by Gaussian fitting. RAD16 is located
240kb from CENII, approximately midway between the centromere and telomere (Figure
1A). To quantify subnuclear confinement, we calculate the radius of confinement (Rc) of
the array from the standard deviation of spot positions, σ, and the average squared
deviation from the mean position, ⟨r2
0⟩, as previously described (Verdaasdonk et al.,
2013).
A wild-type (WT) undamaged RAD16 locus explores an area with Rc = 705 nm
(Figure 1C; Table 1). To investigate the effect of DNA damage, cells were treated with
phleomycin, a DNA intercalating agent that induces DSBs randomly throughout the
genome (Chen, Ghorai, Kenney, & Stubbe, 2008). After treatment with low
concentration of phleomycin, RAD16 mobility significantly increased to Rc = 854nm
(Figure 1C; Table 1), occupying a 78% greater nuclear volume than undamaged cells. To
strain engineered with an I-SceI cleavage site at LYS2, 4.7kb away from RAD16 (Figure
1A). I-SceI induction increased RAD16 mobility to Rc = 1103nm (Figure 1C; Table 1),
exploring a 282% greater nuclear volume than uninduced cells. These data show that a
lacO array on the arm on chromosome 2 increases mobility significantly in response to
many random DSBs as well as a single DSB proximal to the lacO array.
To investigate the global nature of the DDR, we use cohesin distribution during
mitosis as readout of chromatin behavior. In mitosis, cohesin is enriched at the
pericentromere and becomes cylindrically arrayed around the mitotic spindle (Ambrosio
et al., 2008; Eckert, Gravdahl, & Megee, 2007; Li et al., 2011; Ng, Waples, Lavoie, &
Biggins, 2009; Stephens, Haase, Vicci, Taylor, & Bloom, 2011). We measure sagittal
width of GFP-labeled cohesin (Smc3-GFP) to quantitate the physical state of chromatin
(Figure 1D). WT cohesin has a diameter (d) of 408nm (Figure 1E; Table 2). Treatment
with phleomycin significantly expanded the cohesin cylinder to d = 455nm (Figure 1E;
Table 2), increasing its volume by 24%. Induction of a single DSB at the MAT locus via
an HO endonuclease expanded cohesin to d = 505nm (Figure 1E; Table 2), a 53% greater
volume than WT. Thus, induction of either many random DSBs or a single DSB on the
arm of chromosome 3 causes significant expansion of pericentromeric cohesin, providing
* * *
*
A D
B
C
E
Figure 1.
(A) Map of S. cerevisiae strain KS406. lacI-GFP binds to lacO at RAD16, 240kb from the centromere on chromosome 2. I-SceI cut site is at LYS2, 4.7kb away from RAD16.
(B) Example of a cell (KS406) in G1 with lacO/lacI-GFP and Spc29-RFP foci. Scale bar, 1µm. (C) Rc values calculated for lacO/lacI-GFP at RAD16 under DNA damage. Phleomycin treatment and induction of a single DSB significantly increased RAD16 mobility (p < 0.05). Scatter plots are included illustrating variance in lacO/lacI-GFP spot position relative to SPB. Scale bar, 500nm.
(D) Example of a cell (WLY8912/KBY6050) in mitosis with Smc3-GFP and Spc29-RFP in WT and under DNA damage. Scale bar, 1µm.
(E) Cohesin sagittal width under DNA damage. Phleomycin treatment and induction of a single DSB caused significant expansion of cohesin (p < 0.05).
Global response to DNA damage is dependent on DNA checkpoint
Increased mobility of a DSB has been shown to depend on RAD51, RAD54,
MEC1 and RAD9 (Dion et al., 2012, Figure 2A; Table 1). We find, in rad9Δ mutants, the
cohesin cylinder does not significantly expand upon treatment with phleomycin or
both chromatin spot expansion and expansion of the cohesin cylinder upon DNA damage
depends on RAD9.
Microtubule poison has also been shown to increase cohesin cylinder width
(Haase, Stephens, Verdaasdonk, Yeh, & Bloom, 2012). If the mechanisms for cohesin
and chromatin expansion are conserved between the spindle assembly checkpoint and the
DDR, we would predict chromatin mobility to increase upon microtubule poison. To test
this, we used nocodazole to reduce microtubule dynamics (Samson, Donoso,
Heller-Bettinger, Watson, & Himes, 1979) and found RAD16 mobility increased to Rc = 791nm,
similar to the effect upon phleomycin treatment (Figure 2C; Table 1). Thus, the cell
increases chromatin mobility as a response to a variety of perturbations, including both
DNA and spindle damage.
What is the mechanism for increased chromatin mobility?
There are at least 3 possible mechanisms for increased chromatin motion: (1) physical
changes in chromatin, (2) nuclear rocking by the cytoskeleton and (3) changes in the
nuclear environment (i.e. nuclear proteins). If chromatin is physically altered upon
damage, simple polymer theories might account for the increased mobility observed. For
example, elements of the DDR may modify the chromatin polymer, modulating its
persistence length (Lp). Lp is defined as the length over which two spots on a chain lose
directional correlation with each other, and increased Lp causes increased radius of
gyration, a measure of the volume a polymer can occupy (Bloom, 2008). Thus, increased
*
A B
C D
Figure 2.
(A) Rc values calculated for lacO/lacI-GFP at RAD16 in a rad9Δ strain (KBY8819). Phleomycin treatment did not significantly increase mobility (p > 0.05). Scale bar, 500nm.
(B) Cohesin sagittal width in a rad9Δ strain (KBY6054). Neither phleomycin treatment nor induction of a single DSB caused significant expansion of cohesin (p > 0.05).
(C) Rc values calculated for lacO/lacI-GFP at RAD16 under microtubule damage. Nocodazole treatment caused a significant increase in mobility (p < 0.05). Scale bar, 500nm
(D) Cohesin sagittal width in two histone mutants, S129A (KBY6062) and S121A (KBY8785). Phleomycin treatment did not cause significant expansion of cohesin in either mutant (p > 0.05).
We hypothesize chromatin Lp is modulated due to histone phosphorylation by
effector kinases in the DDR. One possible histone candidate is H2A, for expansion of
2012). To test if cohesin expansion upon DNA damage is also dependent on the
phosphorylation of H2A, we measure cohesin sagittal width in two unphosphorylatable
histone mutants, H2A-S121A and H2A-S129A. Upon treatment with phleomycin, the
cohesin barrel did not significantly expand in either mutant (Figure 2D; Table 2). Thus,
expansion of cohesin upon either DNA or spindle damage is dependent on H2A
phosphorylation, evidence for conservation between the spindle and DNA damage
responses.
Nuclear dynamics depend on actin
To investigate whether components of the cytoskeleton modify nuclear motion
upon DNA damage, we quantify SPB (Spc29-RFP) dynamics in addition to RAD16. The
SPB is embedded in the nuclear membrane, and we use the same analysis for Rc as
described earlier. A WT SPB explores an area with Rc = 698nm (Figure 3; Table 1).
Treatment with phleomycin increased SPB mobility to Rc = 782nm, and induction of a
single DSB increased SPB mobility to Rc = 1219nm (Figure 3; Table 1). Thus, SPB
dynamics increase in response to DNA damage, suggesting nuclear dynamics are
enhanced upon DNA damage in addition to chromatin dynamics.
To investigate whether the cytoskeleton drives this response, we used nocodazole
to reduce microtubule dynamics and found no change in SPB mobility (Table 1). Thus,
WT SPB dynamics are independent of microtubules. Next,to test the role of actin in
modulating SPB dynamics, we used latrunculin A to inhibit actin polymerization
Figure 3.
Rc values calculated for lacO/lacI-GFP at RAD16 and SPB under states of actin and/or DNA damage. Single DSB induction significantly increases lacO and SPB mobility. Latrunculin A does not affect lacO but decreases SPB mobility. Bars with different numbers of asterisks are significantly different (p < 0.05). Scale bar, 500nm.
significantly decreased SPB mobility to Rc = 483nm (Figure 3; Table 1) but did not affect
RAD16 mobility, showing nuclear but not chromatin dynamics depend on actin. To investigate whether actin contributes to increased chromatin and/or nuclear
motion upon DNA damage, we induced a single DSB followed by treatment with
latrunculin A and found RAD16 moved with Rc = 763nm and the SPB with Rc = 645,
both intermediate relative to DSB induction or latrunculin A treatment alone (Figure 3
Table 1). Since RAD16 mobility under this condition was greater than latruncuilin A
treatment alone, an actin-independent mechanism for increasing chromatin mobility
exists. However, RAD16 mobility was also less than single DSB alone, indicating an
actin-dependent mechanism for increasing chromatin mobility as well. Fluctuations in
nuclear dynamics directly correlate with lacO mobility, evidence that nuclear dynamics
affect the motion of its contents. Thus, chromatin dynamics can be modulated by altering
to maintain normal nuclear dynamics and achieve increased chromatin mobility upon
DNA damage. Further investigation is needed to determine if actin is activated upon
DNA damage to increase nuclear dynamics.
Table 1. Summary of movement parameters Spot tracked Relevant
genotype
Treatment or damage Rc (nm)
Carbon source
Cell cycle
No. of cells RAD16 (lacI-‐GFP) WT None 705 Glucose G1 43 WT 3µg/mL phleomycin 854 Glucose G1 68 WT single DSB (I-‐SceI) 1103 Galactose G1 14 WT 50µM latrunculin A 703 Glucose G1 15 WT single DSB (I-‐SceI)/
50µM latrunculin A
770 Galactose G1 23
WT 20µg/mL nocodazole 791 Glucose G1 61 rad9 None 1041 Glucose G1 25 rad9 3µg/mL phleomycin 1000 Glucose G1 22 SPB (Spc29-‐RFP) WT None 698 Glucose G1 49 WT 3µg/mL phleomycin 782 Glucose G1 71 WT single DSB (I-‐SceI) 1219 Galactose G1 16 WT 50µM latrunculin A 483 Glucose G1 16 WT single DSB (I-‐SceI)/
50µM latrunculin A
645 Galactose G1 23
WT 20µg/mL nocodazole 698 Glucose G1 65 Nucleus (Nup49-‐GFP) WT None 485 Glucose G1 14 WT 50µM latrunculin A Glucose G1 11
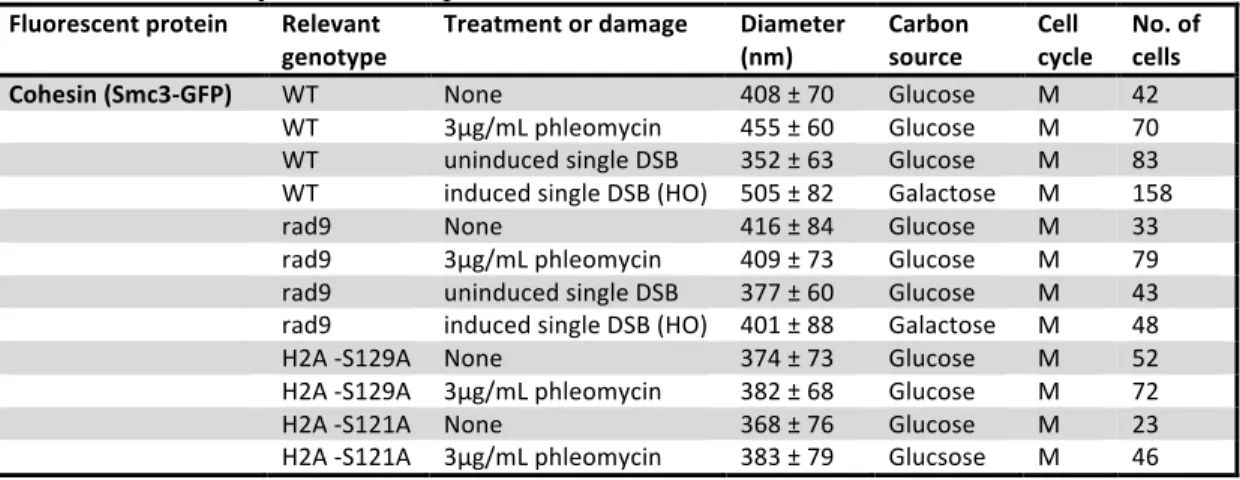
Table 2. Summary of cohesin parameters Fluorescent protein Relevant
genotype
Treatment or damage Diameter (nm)
Carbon source
Cell cycle
DISCUSSION
How do you increase chromatin motion?
Chromatin exhibits increased mobility upon DSB induction, but the mechanism
remains unknown. It might be that the loose ends roam freely; however, it has been
shown that broken ends of a DSB remain in close proximity (Kaye et al., 2004;
Lobachev, Vitriol, Stemple, Resnick, & Bloom, 2004). Also, the response is global, for it
has been shown that an uncut chromosome increases exploration upon induction of a
DSB on its homolog (Miné-Hattab & Rothstein, 2012). Our work further characterizes
this global response. We now consider three possible mechanisms for the global increase
in chromatin mobility upon DNA damage: (1) altered enzyme activity, (2) change in Lp
and (3) the cytoskeleton.
Chromatin mobility is sensitive to ATP levels (Weber, Spakowitz, & Theriot,
2012), but it is not completely clear why. Most likely, this sensitivity results from the
ATP-dependent activity of chromatin remodelers. For example, removal of nucleosomes
at the PHO5 promoter region by the two remodeling complexes Swi2/Snf2 and INO80
increases PHO5 mobility (Barbaric et al., 2007; Neumann et al., 2012; Steger, Haswell,
Miller, Wente, & O’Shea, 2003). Interestingly, targeting an undamaged locus with
INO80 has also been shown to increase mobility at that locus (Neumann et al., 2012).
With this evidence, others have proposed chromatin-remodeling activities can be used to
promote recombination and are regulated by the DNA repair machinery and the DDR
(Dion & Gasser, 2013).This is plausible, but not likely, as it is difficult to imagine the
Chromatin can be modeled as a bead-spring chain doubly tethered at both ends
with the parameters temperature (T), contour length (Lc), compaction ratio (Cr) and Lp
(Verdaasdonk et al., 2013). Altering these parameters can change how dynamic the
polymer is and can help explain why chromatin mobility is increased upon DNA damage.
It is unlikely that T or Lc change in response to DNA damage, but alterations in Cr or Lp
are possible. As stated earlier, increasing Lp causes an increase in radius of gyration, a
measure of the volume a polymer can explore (Bloom, 2008). This effect results from
increased stiffness of the chain, as the distance over which the polymer can bend
increases with Lp. Thus, less “wiggle room” is available to the polymer when Lp
increases, and it occupies a greater volume. Therefore, an increase in Lp of chromatin
could be responsible for increased chromatin mobility observed upon induction of DSBs.
We predict the cell modulates Lp in response to DNA damage via checkpoint
proteins, as we have shown cohesin expansion upon DSB induction is dependent on
phosphorylation of histone by checkpoint kinases (Figure 2B and 2D; Table 2). The
phosphorylation sites we tested, S121 and S129, are found at the C-terminal tail of
histone H2A. Bub1 phosphorylates S121 in response to spindle damage (Haase et al.,
2012; Kawashima, Yamagishi, Honda, Ishiguro, & Watanabe, 2010); Mec1 and Tel1
phosphorylate S129 in response to DNA damage (Melo, Cohen, & Toczyski, 2001;
Nakada & Matsumoto, 2003; Zou & Elledge, 2003). Thus, the spindle checkpoint and
DDR both modulate chromatin through phosphorylation at the C-terminus of H2A upon
damage. Interestingly, these residues are found near the site where the DNA enters and
exits the nucleosome (White, Suto, & Luger, 2001). We hypothesize adding a highly
around the nucleosome, physically altering the chromatin polymer and effectively
increasing Lp. This would explain why cohesin expansion and increased chromatin
mobility are observed upon either spindle or DNA damage.
The cytoskeleton is dynamic and could be utilized to alter the nuclear
environment. For example, upon DNA damage, the cytoskeleton could cause rocking of
the nucleus, mixing its contents and, thus, promoting homology search. In budding yeast,
actin has been implicated to alter nuclear dynamics in meiosis, another instance when
homology search is key (Koszul et al., 2008; Scherthan et al., 2007; Trelles-Sticken et al.,
2005). We have shown that inhibiting microtubule polymerization has no effect on
nuclear motion, but that inhibiting actin reduces nuclear dynamics in WT or after DSB
induction. Thus, nuclear dynamics rely on actin and, theoretically, could be altered in an
actin-dependent manner. Further investigation is needed to determine whether the cell
utilizes this mechanism in response to DNA damage to promote nuclear mixing and
facilitate homology search.
EXPERIMENTAL PROCEDURES
The budding yeast Saccharomyces cerevisiae served as the model organism for
this study. For detailed growth and imaging conditions, see Supplemental Experimental
Procedures. Strain genetic backgrounds can be found in Table S1.
Image Analysis
Images were analyzed using MetaMorph (Molecular Devices, Sunnyvale) and
GFP or RFP pixel out of each seven plane stack was used to determine the coordinates of
a tagged spot at that time point. To track a lacO or SPB spot, we use a custom MATLAB
program (Speckle Tracker) as previously described (Wan, 2008; Wan et al., 2009; Wan,
Cimini, Cameron, & Salmon, 2012) to perform Gaussian fitting and achieve subpixel
centroid mapping. To track the center of the nucleus, we use Integrated Morphometry
Analysis in MetaMorph to determine the center of the fluorescent Nup49-GFP.
Coordinates were further analyzed using MATLAB and Excel (Microsoft, Redmond).
For chromatin array Rc measurements, Spc29-RFP coordinates were subtracted from
lacO/lacI-GFP coordinates to control for cellular and nuclear motion. To measure cohesin
sagittal width, we use a Linescan in MetaMorph to determine the distance between
brightest pixels on opposite sides of the cylinder.
Calculating Rc from Experimental Data
MATLAB was used to fit the spot positions as µμ!,σ! = normfit(x−x!"#$)
and µμ!,σ! = normfit(y−y!"#$). The variance of the distribution of spot positions is
then calculated as σ2 = mean(σ2x, σ2y). The average squared deviation from the mean
position is ∆r!! = ∆x
!
! + ∆y
!! . Using σ! and ∆r!! , we calculate Rc as
R! =5
4∗ 2σ!+ ∆r!!
Statistical Analysis
For statistical comparison of reported Rc values, we used Levene’s test to compare
homogeneity of population variances. To compare cohesin sagittal width measurements,
we used a Student’s t-test (p values found in Table S2).
REFERENCES
Agarwal, S., Tafel, A. A., & Kanaar, R. (2006). DNA double-strand break repair and
chromosome translocations. DNA Repair, 5(9-10), 1075–1081.
doi:10.1016/j.dnarep.2006.05.029
Ambrosio, C. D., Schmidt, C. K., Katou, Y., Kelly, G., Itoh, T., Shirahige, K., & Uhlmann, F. (2008). Identification of cis-acting sites for condensin loading onto
budding yeast chromosomes. Genes & Development, 22(16), 2215–2227.
doi:10.1101/gad.1675708.Freely
Barbaric, S., Luckenbach, T., Schmid, A., Blaschke, D., Hörz, W., & Korber, P. (2007). Redundancy of chromatin remodeling pathways for the induction of the yeast PHO5
promoter in vivo. The Journal of Biological Chemistry, 282(38), 27610–27621.
doi:10.1074/jbc.M700623200
Bloom, K. S. (2008). Beyond the code: the mechanical properties of DNA as they relate
to mitosis. Chromosoma, 117(2), 103–110. doi:10.1007/s00412-007-0138-0.Beyond
Chen, J., Ghorai, M. K., Kenney, G., & Stubbe, J. (2008). Mechanistic studies on bleomycin-mediated DNA damage: multiple binding modes can result in
double-stranded DNA cleavage. Nucleic Acids Research, 36(11), 3781–3790.
doi:10.1093/nar/gkn302
Chikashige, Y., Ding, D.-Q., Funabiki, H., Haraguchi, T., Mashiko, S., Yanagida, M., & Hiraoka, Y. (1994). Telomere-led premeiotic chromosome movement in fission
yeast. Science, 264(5156), 270–273. Retrieved from
http://www.sciencemag.org/content/264/5156/270.full.pdf
Dion, V., & Gasser, S. M. (2013). Chromatin movement in the maintenance of genome
stability. Cell, 152(6), 1355–1364. doi:10.1016/j.cell.2013.02.010
recombination machinery. Nature Cell Biology, 14(5), 502–509. doi:10.1038/ncb2465
Eckert, C. A., Gravdahl, D. J., & Megee, P. C. (2007). The enhancement of
pericentromeric cohesin association by conserved kinetochore components promotes high-fidelity chromosome segregation and is sensitive to microtubule-based tension.
Genes & Development, 21(3), 278–291. doi:10.1101/gad.1498707.somes
Gehlen, L. R., Gasser, S. M., & Dion, V. (2011). How Broken DNA Finds Its Template
for Repair: A Computational Approach. Progress of Theoretical Physics
Supplement, 191(191), 20–29. doi:10.1143/PTPS.191.20
Haase, J., Stephens, A., Verdaasdonk, J., Yeh, E., & Bloom, K. (2012). Bub1 kinase and Sgo1 modulate pericentric chromatin in response to altered microtubule dynamics.
Current Biology, 22(6), 471–481. doi:10.1016/j.cub.2012.02.006
Kawashima, S. A., Yamagishi, Y., Honda, T., Ishiguro, K., & Watanabe, Y. (2010). Phosphorylation of H2A by Bub1 prevents chromosomal instability through
localizing shugoshin. Science, 327(5962), 172–177. doi:10.1126/science.1180189
Kaye, J. A., Melo, J. A., Cheung, S. K., Vaze, M. B., Haber, J. E., & Toczyski, D. P. (2004). DNA breaks promote genomic instability by impeding proper chromosome
segregation. Current Biology, 14(23), 2096–2106. doi:10.1016/j
Koszul, R., Kim, K., Prentiss, M., Kleckner, N., & Kameoka, S. (2008). Meiotic chromosomes move by linkage to dynamic actin cables with transduction of force
through the nuclear envelope. Cell, 133(7), 1188–1201.
doi:10.1016/j.cell.2008.04.050
Lee, C.-Y., Conrad, M. N., & Dresser, M. E. (2012). Meiotic chromosome pairing is promoted by telomere-led chromosome movements independent of bouquet
formation. PLoS Genetics, 8(5), e1002730. doi:10.1371/journal.pgen.1002730
Li, Z., Vizeacoumar, F. J., Bahr, S., Li, J., Warringer, J., Vizeacoumar, F. S., … Boone, C. (2011). Systematic exploration of essential yeast gene function with
temperature-sensitive mutants. Nature Biotechnology, 29(4), 361–367. doi:10.1038/nbt.1832
Lobachev, K., Vitriol, E., Stemple, J., Resnick, M. A., & Bloom, K. (2004). Chromosome Fragmentation after Induction of a Double-Strand Break Is an Active Process
Prevented by the RMX Repair Complex. Current Biology, 14(23), 2107–2112.
doi:10.1016/j
Maguire, M. P. (1974). A New Model for Homologous Chromosome Pairing.
Melo, J. A., Cohen, J., & Toczyski, D. P. (2001). Two checkpoint complexes are
independently recruited to sites of DNA damage in vivo. Genes & Development,
15(21), 2809–2821. doi:10.1101/gad.903501.Weinert
Miné-Hattab, J., & Rothstein, R. (2012). Increased chromosome mobility facilitates
homology search during recombination. Nature Cell Biology, 14(5), 510–517.
doi:10.1038/ncb2472
Nakada, D., & Matsumoto, K. (2003). ATM-related Tel1 associates with double-strand
breaks through an Xrs2-dependent mechanism. Genes & Development, 17(16),
1957–1962. doi:10.1101/gad.1099003.may
Neumann, F. R., Dion, V., Gehlen, L. R., Tsai-Pflugfelder, M., Schmid, R., Taddei, A., & Gasser, S. M. (2012). Targeted INO80 enhances subnuclear chromatin movement
and ectopic homologous recombination. Genes & Development, 26(4), 369–383.
doi:10.1101/gad.176156.111
Ng, T. M., Waples, W. G., Lavoie, B. D., & Biggins, S. (2009). Kinetochore
Biorientation. Molecular Biology of the Cell, 20(17), 3818–3827.
doi:10.1091/mbc.E09
Rassool, F. V. (2003). DNA double strand breaks (DSB) and non-homologous end
joining (NHEJ) pathways in human leukemia. Cancer Letters, 193(1), 1–9.
Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/12691817
Samson, F., Donoso, J. A., Heller-Bettinger, I., Watson, D., & Himes, R. H. (1979). Nocodazole Action and Fast Axoplasmic on Tubulin Assembly, Axonal
Ultrastructure and Fast Axoplasmic Transport. The Journal of Pharmacology and
Experimental Therapeutics, 208(3), 411–417.
Savage, J. R. (1996). Insight into sites. Mutation Research, 366(2), 81–95. Retrieved
from http://www.ncbi.nlm.nih.gov/pubmed/9001576
Scherthan, H., Wang, H., Adelfalk, C., White, E. J., Cowan, C., Cande, W. Z., & Kaback, D. B. (2007). Chromosome mobility during meiotic prophase in Saccharomyces
cerevisiae. PNAS, 104(43), 16934–16939. doi:10.1073/pnas.0704860104
Spector, I., Shochet, N. R., Blasberger, D., & Kashman, Y. (1989). Latrunculins--novel marine macrolides that disrupt microfilament organization and affect cell growth: I.
Comparison with cytochalasin D. Cell Motility and the Cytoskeleton, 13(3), 127–
144. doi:10.1002/cm.970130302
Steger, D. J., Haswell, E. S., Miller, A. L., Wente, S. R., & O’Shea, E. K. (2003).
Regulation of chromatin remodeling by inositol polyphosphates. Science,
Stephens, A. D., Haase, J., Vicci, L., Taylor, R. M., & Bloom, K. (2011). Cohesin, condensin, and the intramolecular centromere loop together generate the mitotic
chromatin spring. The Journal of Cell Biology, 193(7), 1167–1180.
doi:10.1083/jcb.201103138
Thorslund, T., & West, S. C. (2007). BRCA2: a universal recombinase regulator.
Oncogene, 26(56), 7720–7730. doi:10.1038/sj.onc.1210870
Trelles-Sticken, E., Adelfalk, C., Loidl, J., & Scherthan, H. (2005). Meiotic telomere
clustering requires actin for its formation and cohesin for its resolution. The Journal
of Cell Biology, 170(2), 213–223. doi:10.1083/jcb.200501042
Verdaasdonk, J. S., Vasquez, P. A., Barry, R. M., Barry, T., Goodwin, S., Forest, M. G., & Bloom, K. (2013). Centromere tethering confines chromosome domains.
Molecular Cell, 52(6), 819–831. doi:10.1016/j.molcel.2013.10.021
Wan, X. (2008). Asymmetric Chromosome Oscillation during Mitosis and Protein
Architecture of the Human Kinetochore Measured by K-SHREC (Kinetochore-Speckle High Resolution Co-Localization). PhD thesis, University of North Carolina at Chapel Hill, Chapel Hill, NC. http://dc.lib.unc.edu/cdm/ref/collection/etd/id/2050
Wan, X., Cimini, D., Cameron, L. a, & Salmon, E. D. (2012). The coupling between sister kinetochore directional instability and oscillations in centromere stretch in
metaphase PtK1 cells. Molecular Biology of the Cell, 23(6), 1035–1046.
doi:10.1091/mbc.E11-09-0767
Wan, X., O’Quinn, R. P., Pierce, H. L., Joglekar, A. P., Gall, W. E., DeLuca, J. G., … Salmon, E. D. (2009). Protein architecture of the human kinetochore microtubule
attachment site. Cell, 137(4), 672–684. doi:10.1016/j.cell.2009.03.035
Weber, S. C., Spakowitz, A. J., & Theriot, J. a. (2012). Nonthermal ATP-dependent
fluctuations contribute to the in vivo motion of chromosomal loci. PNAS, 109(19),
7338–7343. doi:10.1073/pnas.1119505109
White, C. L., Suto, R. K., & Luger, K. (2001). Structure of the yeast nucleosome core
particle reveals fundamental changes in internucleosome interactions. The EMBO
Journal, 20(18), 5207–5218. doi:10.1093/emboj/20.18.5207
Wyman, C., & Kanaar, R. (2006). DNA double-strand break repair: all’s well that ends
well. Annual Review of Genetics, 40, 363–383.
doi:10.1146/annurev.genet.40.110405.090451
Zou, L., & Elledge, S. J. (2003). Sensing DNA damage through ATRIP recognition of
RPA-ssDNA complexes. Science, 300(5625), 1542–1548.
SUPPLEMENTAL EXPERIMENTAL PROCEDURES
Cell preparations
Genotypes can be found in Table S1. Cells were grown overnight in the proper
media, usually YPD, at 24° C. These overnight cultures were diluted the next morning, a
few hours prior to imaging, to obtain logarithmic phase growth. To induce many DSBs,
cells were treated with 3ug/mL phleomycin for 30 min. To induce a single DSB at a
known locus, cells were grown on YPG for approximately 5 hrs prior to imaging to
induce the I-SceI or HO endonuclease. To inhibit microtubule polymerization, cells were
treated with 20ug/mL nocodazole for 1 hr. To inhibit actin polymerization, cells were
treated with 50µM latrunculin A for 15 min. For imaging, cells were washed and
resuspended in YC complete media with 2% glucose (or 2% galactose for activation of a
Gal-promoter).
Microscopy:
Images were acquired using a Nikon Eclipse Ti wide-field inverted microscope
with a 100x Apo TIRF 1.49 NA objective (Nikon, Melville, New York, USA) and Andor
Clara CCD camera (Andor, South Windsor, Connecticut, USA). Three-dimensional
stacks, each compiled of seven 200nm z planes, were acquired at room temperature every
30s over a 10 minute period (147 total planes per time-lapse) with Nikon NIS Elements
imaging software (Nikon, Melville, New York, USA). Images were taken in Trans, GFP,
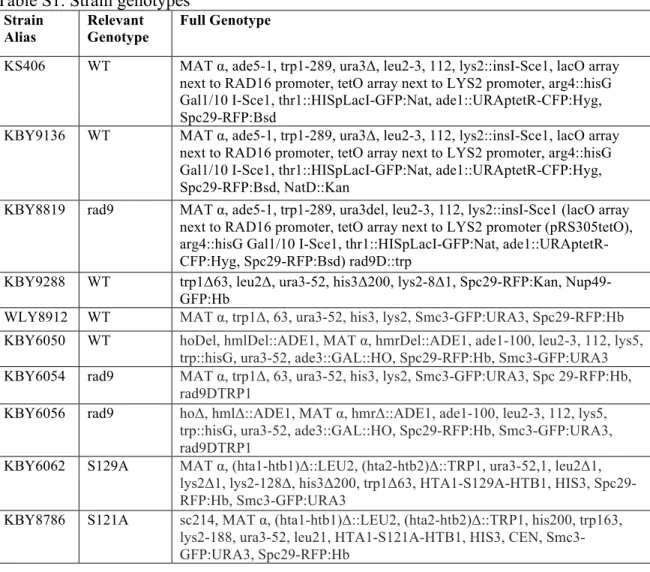
Table S1. Strain genotypes
Strain Alias
Relevant Genotype
Full Genotype
KS406 WT MAT α, ade5-1, trp1-289, ura3Δ, leu2-3, 112, lys2::insI-Sce1, lacO array next to RAD16 promoter, tetO array next to LYS2 promoter, arg4::hisG Gal1/10 I-Sce1, thr1::HISpLacI-GFP:Nat, ade1::URAptetR-CFP:Hyg, Spc29-RFP:Bsd
KBY9136 WT MAT α, ade5-1, trp1-289, ura3Δ, leu2-3, 112, lys2::insI-Sce1, lacO array next to RAD16 promoter, tetO array next to LYS2 promoter, arg4::hisG Gal1/10 I-Sce1, thr1::HISpLacI-GFP:Nat, ade1::URAptetR-CFP:Hyg, Spc29-RFP:Bsd, NatD::Kan
KBY8819 rad9 MAT α, ade5-1, trp1-289, ura3del, leu2-3, 112, lys2::insI-Sce1 (lacO array next to RAD16 promoter, tetO array next to LYS2 promoter (pRS305tetO), arg4::hisG Gal1/10 I-Sce1, thr1::HISpLacI-GFP:Nat, ade1::URAptetR-CFP:Hyg, Spc29-RFP:Bsd) rad9D::trp
KBY9288 WT trp1Δ63, leu2Δ, ura3-52, his3Δ200, lys2-8Δ1, Spc29-RFP:Kan, Nup49-GFP:Hb
WLY8912 WT MAT α, trp1Δ, 63, ura3-52, his3, lys2, Smc3-GFP:URA3, Spc29-RFP:Hb
KBY6050 WT hoDel, hmlDel::ADE1, MAT α, hmrDel::ADE1, ade1-100, leu2-3, 112, lys5, trp::hisG, ura3-52, ade3::GAL::HO, Spc29-RFP:Hb, Smc3-GFP:URA3
KBY6054 rad9 MAT α, trp1Δ, 63, ura3-52, his3, lys2, Smc3-GFP:URA3, Spc 29-RFP:Hb, rad9DTRP1
KBY6056 rad9 hoΔ, hmlΔ::ADE1, MAT α, hmrΔ::ADE1, ade1-100, leu2-3, 112, lys5, trp::hisG, ura3-52, ade3::GAL::HO, Spc29-RFP:Hb, Smc3-GFP:URA3, rad9DTRP1
KBY6062 S129A MAT α, (hta1-htb1)Δ::LEU2, (hta2-htb2)Δ::TRP1, ura3-52,1, leu2Δ1, lys2Δ1, lys2-128Δ, his3Δ200, trp1Δ63, HTA1-S129A-HTB1, HIS3, Spc29-RFP:Hb, Smc3-GFP:URA3
Table S2. p values using Levene’s test for Rc and Student’s t-test for cohesin diameter
RAD16 Rc
Levene's Test (p<0.05) +phleomycin +single DSB +latrunculin A +DSB +latA +nocodazole WT 4.87E-‐10 6.40E-‐29 9.17E-‐01 5.30E-‐03 5.10E-‐05
rad9 3.94E-‐01
SPB Rc
Levene's Test (p<0.05) +phleomycin +single DSB +latrunculin A +DSB +latA +nocodazole WT 7.91E-‐04 4.58E-‐35 4.33E-‐11 4.82E-‐02 9.91E-‐01 rad9 2.70E-‐03
Cohesin Diameter
Student's T Test (p<0.05) +phleomycin +single DSB
WT 3.29E-‐04 6.15E-‐35
rad9 1.47E-‐01
S129A 5.22E-‐01
S121A 4.40E-‐01