Abstract
Platelets play a critical role in the body’s response to vascular injury (hemostasis), but they also
contribute significantly to blood coagulation (thrombosis) associated with cardiovascular
disease. The small GTPase RAP1 in platelets is an essential component of the thrombotic
pathway as it controls the adhesive state of platelets in circulation and at sites of vascular injury.
The guanine nucleotide exchange factor, CalDAG-GEFI, and the GTPase-activating protein,
RASA3, are key regulators of RAP1 and balance its activation state. Mice that are deficient in
CalDAG-GEFI exhibit abnormal bleeding due to impaired platelet activation and adhesion. Mice
lacking RASA3 are thrombocytopenic (low platelet count) due to premature platelet activation
and clearance. We hypothesize that interindividual variability in the antagonistic balance
between CalDAG-GEFI and RASA3 predicts the risk for thrombosis or
thrombocytopenia-induced bleeding. This study aims to establish an assay for the rapid quantification of
CalDAG-GEFI and RASA3 expression in platelets. We performed flow cytometric quantification of
intracellular immunofluorescence signals to measure CalDAG-GEFI and RASA3 expression in
mouse platelets that were wild-type, heterozygous, or null for RASA3 or CalDAG-GEFI. In
parallel, we quantified CalDAG-GEFI and RASA3 expression in these platelets by Western
blotting, which can be used to assay protein levels as well but is time-consuming and not
currently viable as a clinical test. Our results validate flow cytometry as a powerful technique to
quantify RASA3 and CalDAG-GEFI expression in mouse platelets, as results obtained by flow
cytometry strongly correlated with those obtained by Western blotting. These results indicate the
potential viability of flow cytometry as a rapid clinical tool to identify the biochemical basis of
Introduction
Platelets are key players in the hemostatic process. These small anucleated blood cells
have numerous biological functions, from the inflammatory response to promoting tumor
angiogenesis[1], but one of their most prominent functions is in the body’s response to vascular
damage. At the site of an injury, they activate, adhere to the subendothelial extracellular matrix,
form a platelet plug, and put out a fibrin mesh to strongly coagulate the blood and trap other cell
types in the plug[1].
This study focuses on platelet activation, which is governed primarily by the RAP1
activation pathway[3]. RAP1 is a small GTPase in the subfamily of Ras-related proteins; it has
two isoforms, RAP1A and RAP1B[3]. Its activation in platelets results most notably in the
activation of the integrin αIIbβ3, which binds to extracellular fibrinogen and results in
platelet-platelet adhesion and the formation of the initial platelet-platelet plug[1]. Upstream of αIIbβ3 is talin,
which was knocked out of some mice (via Cre-Lox recombination) in this experiment to allow
experimentation on mice that are deficient in the GTPase-activating protein (GAP) RASA3.
RASA3 knockout mice are embryonic lethal[4], so these mice and HLB mutant mice (who are
also RASA3-deficient) had to be used as a substitute.
RAP1, as a small GTPase, cycles between an inactive GDP-bound form and an active
GTP-bound form[4]. Activation is governed by the guanine exchange factor CalDAG-GEFI, which
itself is dependent on Ca2+ and diacylglycerol[6]. CalDAG-GEFI promotes GTP loading into both
isoforms of RAP1 as part of a rapid activation pathway[1]. Slower, more sustained activation
pathways include signaling via PKC, P2Y12, and PI3K[4]. RAP1 inactivation has recently been
implicated to be sustained by RASA3[4]. GAPs catalyze GTP hydrolysis, and RASA3 performs
this function on RAP1 isoforms.
Thus, RAP1 (and by extension, platelet) activation and inactivation are maintained in a
delicate balance primarily by CalDAG-GEFI and RASA3. When one or the other is missing,
aggregation and forming platelet plugs, usually accompanied by bleeding disorders[6]. The
inability of RASA3-deficient mice to completely inactivate their platelets results in heightened
platelet clearance rates, severe thrombosis and thrombocytopenia, and embryonic lethality[4].
The RASA3 pathway in effect restrains the CalDAG-GEFI pathway, preventing platelet
activation until the CalDAG-GEFI pathway overwhelms the RASA3 pathway at a site of vascular
injury. The slow, sustained pathways then maintain the formation of the platelet plug.
The balance of CalDAG-GEFI and RASA3 is as such responsible for the maintenance of
the hemostatic pathway in biological systems. The necessity of the proteins to be expressed at
comparable levels is relevant in platelet-mediated bleeding disorders, and an accessible method
for determining this ratio may be useful in implicating deficiencies in either protein in such
disorders. It is such a method that this paper aims to establish.
Materials and Methods
Mice
Mice of the following genotypes were generated for the purpose of this experiment:
CalDAG-GEFI: +/+, +/-, -/- generated by Bergmeier et al.2
RASA3: +/+, HLB +/-, Talin fl/fl Cre+ HLB -/- (HLB mutants are deficient in RASA3). HLB mice
generated by Peters et al.4; Talin fl/fl mice generated by Petrich et al.5
P2Y12 -/- mice generated by Pfizer.
Various genotypes possible from combining the two genes (e.g. CalDAG-GEFI +/+, RASA3 +/+;
CalDAG-GEFI +/-, RASA3 +/+, etc.) were bred in-house. The P2Y12 phenotype was not
relevant to the experiment.
Platelet Preparation (Final Protocol)
Approximately 50 μL of blood was collected using heparin-coated capillary tubes from the
phosphate-buffered saline (DPBS 1X, Gibco) + 30 IU/mL heparin. Paraformaldehyde (1%, 900 μL; stock
32% from Electron Microscopy Sciences) + 5% EDTA (solid stock EDTA from Fisher Scientific)
was added to each tube, and the samples were allowed to fix on a rocker for 30 minutes. The
samples were then centrifuged on a hanging bucket centrifuge (Eppendorf Centrifuge 5810 R)
at 800 g for 10 minutes. The supernatant was removed from the samples and the pellet was
resuspended in 500 μL of 0.1% Triton detergent (stock 100X from Fisher Bioreagents). PBS
controls were also made. The samples were allowed to fix for 3 minutes, then 500 μL of PBS
was added to dilute the Triton and stop the permeabilization. The samples were then
centrifuged again at 800 g for 10 minutes. The supernatant was again removed from the
samples and the pellet was resuspended in 500 μL of PBS + 0.01% azide (solid sodium azide
from MP Biochemicals) + 1% bovine serum albumin (solid BSA from US Biological Life
Sciences). The samples were stored at 4ºC.
Flow Cytometry
The fixed and permeabilized samples (and the controls) were labeled with the following
antibodies: anti-CalDAG-GEFI (polyclonal, Thermo Fisher Scientific), anti-RASA3 (polyclonal,
Thermo Fisher Scientific and later polyclonal, Abcam), and anti-GPIB (clone ALMA.16, BD
Biosciences) as a platelet marker. Prior assays also made use of anti-GPIX antibody (clone
Xia.B4, Emfret Analytics) as a platelet marker. A 20-μL reaction mixture was utilized with 5 μL of
sample, 4 μL of a 5X antibody mix, and 11 μL of PBS. The final concentration of the
CalDAG-GEFI antibody was 2 μg/mL, the RASA3 antibody was 10 μg/mL, and the
anti-GPIB antibody was 2 μg/mL. Experiments involving the anti-GPIX antibody used it at a final
concentration of 4 μg/mL. The samples were allowed to hybridize for 30 minutes, then diluted
with 500 μL of PBS. The platelet events were visualized with a BD Accuri C6 Flow Cytometer—
mean fluorescence values for each antibody were observed (CalDAG-GEFI at 488 nm, RASA3
Platelet Lysis for Immunoblot
Approximately 700 μL of blood was collected using heparin-coated capillary tubes from the
retro-orbital plexus of each mouse and was deposited into a tube containing 300 μL of
phosphate-buffered saline (DPBS 1X, Gibco) + 300 IU/mL heparin. The samples were
centrifuged at 120 g for 4 minutes. The platelet-rich plasma (PRP) supernatant was isolated.
The pellet was resuspended in 300 μL of Tyrode’s buffer (137 mM NaCl, 0.3 mM Na2HPO4, 2
mM KCl, 12 mM NaHCO3, 5 mM HEPES, 5 mM dextrose) and centrifuged with the same
parameters. The previous two steps were repeated once more, and the collected PRP was
pooled and centrifuged at 110 g for 5 minutes. The PRP was isolated and centrifuged again at
110 g for 7 minutes. The supernatant was then removed, and 1 μL of prostaglandin I2
(Sigma-Aldrich) was added to it. This solution was then centrifuged at 700 g for 5 minutes. The resultant
supernatant was discarded, and the pellet was resuspended in 100 μL Tyrode’s buffer. The
platelet concentrations were measured and normalized via flow cytometry and subsequent
dilution in cell lysis buffer (20 mM Tris base, 1% Triton-X100, 50 mM NaCl, 250 mM sucrose, 50
mM NaF, 5 mM Na4P2O7). The samples were stored at -20 ºC.
Immunoblot Analysis
The platelet lysates were run on an 4-20% polyacrylamide gel under reducing conditions. The
proteins were transferred onto a polyvinylidene fluoride membrane (Millipore). The following
antibodies were allowed to hybridize to the proteins: anti-CalDAG-GEFI (rabbit polyclonal,
Thermo Fisher Scientific) and anti-RASA3 (goat polyclonal, Santa Cruz Biotechnology). The
membrane was blocked with PBS + 0.2% Tween. The following secondary antibodies were then
allowed to hybridize to the primary antibodies: donkey anti-goat (Alexa Fluor 680, Invitrogen)
and goat anti-rabbit (Dylight 800, Invitrogen). Beta-actin was used as a housekeeping protein,
Sigma-Aldrich) and secondary goat anti-mouse (Dylight 680, Thermo Scientific). The blot was
visualized with a LICOR Odyssey scanner.
Results
The final protocol of the methods section was used to determine CalDAG-GEFI to RASA3 ratios
in a variety of mice. The protocol was refined over the course of the experiment. Preliminary
hybridizations (Figures 1 and 2) were performed to assess the viability of a flow
cytometry-based assay to determine relative expressions of CalDAG-GEFI and RASA3.
Once it was affirmed that CalDAG-GEFI and RASA3 expression could be visualized with some
precision using flow cytometry, the decision was made to use whole blood in the experiments as
opposed to platelet-rich plasma (Figure 3). While whole blood may have contained more
material, thus possibly increasing background fluorescence and masking signal from platelets, it
was ultimately shown to produce more distinct results than PRP, possibly due to cell loss from
manipulations required to extract the PRP.
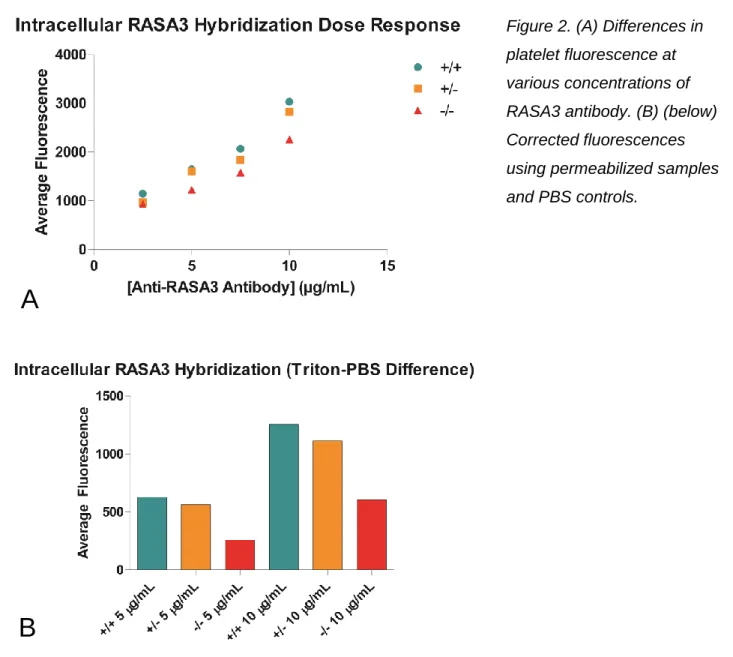
Figure 2. (A) Differences in platelet fluorescence at various concentrations of RASA3 antibody. (B) (below) Corrected fluorescences using permeabilized samples and PBS controls.
A
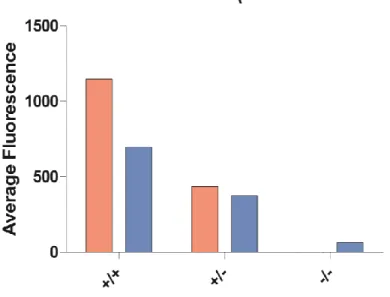
Figure 3. Comparison of fluorescences in whole blood and platelet-rich plasma for CalDAG-GEFI wild-type, heterozygous, and knockout mice. Fluorescence values adjusted using PBS controls.
Next, the optimal detergent and fixative concentration were determined (Figure 4). Triton and
Saponin were tested, and while Saponin was a gentler detergent (thus decreasing damage to
platelet morphology and ideally producing results that were closer to biological conditions),
Triton was found to produce more distinct fluorescence. In the same experiment,
paraformaldehyde concentrations were tested. High concentrations of paraformaldehyde used
during fixation produced methylene bridges and caused antigen masking, so this experiment
was also used to determine the minimum concentration of fixative necessary. The same
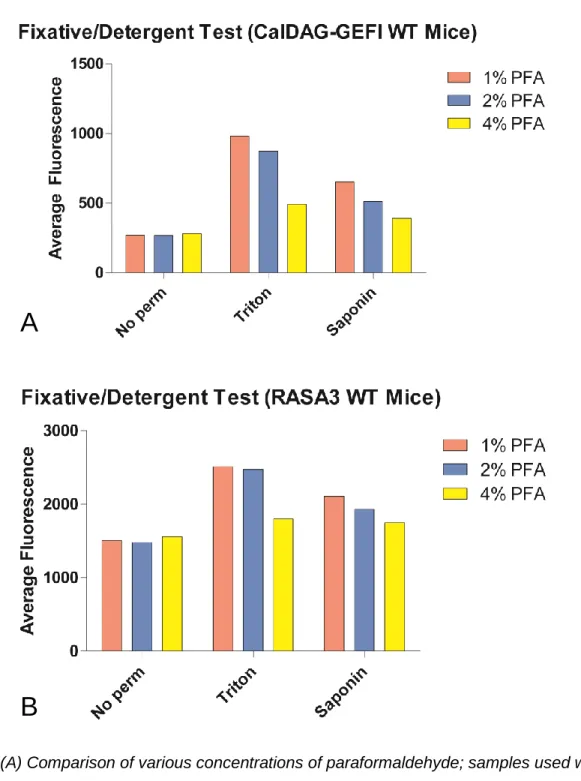
Figure 4. (A) Comparison of various concentrations of paraformaldehyde; samples used were not permeabilized (control) or permeabilized with one of Triton or Saponin. (B) The same procedure, hybridizing with RASA3 rather than CalDAG-GEFI.
Once Triton had been selected as the detergent of choice, the concentration of Triton to be
used had to be determined to assess what level of permeabilization led to the highest
fluorescence while maintaining platelet morphological integrity. Again, the same test was
A
performed using both anti-CalDAG-GEFI antibodies and anti-RASA3 antibodies to compare
fluorescence differences among wild types, heterozygotes, and knockouts within each gene
(Figure 5). The final concentration of Triton to be used was determined to be 0.1%.
Figure 5. (A) CalDAG-GEFI hybridization performed on mice that were wild-type, heterozygous in CalDAG-GEFI and RASA3, and knockouts in both genes. Controls as well as samples treated with 0.1% and 0.5% Triton were compared. (B) The same procedure, hybridizing with CalDAG-GEFI rather than RASA3.
A
In order to check the levels of nonspecific binding of the anti-RASA3 antibody, the hybridized
samples were washed (centrifuged, the supernatant was removed, and the pellet was
resuspended in PBS) twice before running them on the flow cytometer (Figure 6).
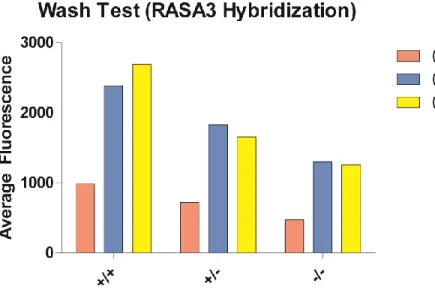
Figure 6. RASA3 hybridization at two concentrations of Triton (and a control) for the same genotypes as in Figure 5.
Once the RASA3 hybridization assay was determined to be reasonably accurate (as seen by
the above figures), fixed and permeabilized blood samples were hybridized with
anti-CalDAG-GEFI and anti-RASA3 antibodies simultaneously (Figure 7), with an anti-GPIB antibody now
being used as a platelet marker rather than the anti-GPIX antibody that was being used
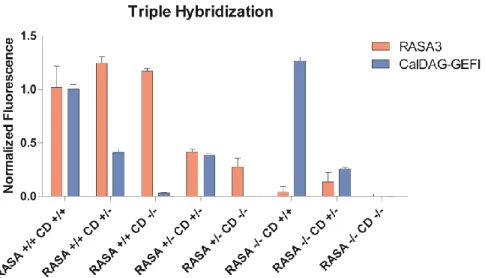
Figure 7. CalDAG-GEFI and RASA3 hybridization with GPIB used as a platelet marker. Values all normalized to the wild-type mean fluorescences. Each genotype had n = 2 or n =3.
After confirming the accuracy of the flow cytometric protocol, it was compared with Western
blotting. CalDAG-GEFI and RASA3 expression were measured (data not shown), and the data
were normalized as a fraction of the wild-type values. The results for both techniques were then
analyzed via linear regression (Figure 8).
Discussion
This study affirms that CalDAG-GEFI to RASA3 ratios can be detected using flow
cytometry. The various parameters tested in this experiment were sufficient to distinguish
RASA3 levels in the different mouse genotypes tested. Anti-RASA3 antibody concentrations of
10 μg/mL were shown to display sufficient levels of fluorescence in the flow cytometer tested
(Figure 2A). In addition, there is a decrease in fluorescence in RASA3 expression in the
non-permeabilized controls (Figure 2B), which is a trend that also presents itself in subsequent
experiments. This can be attributed to the fact that the platelet manipulations (fixation,
centrifugation) could compromise the integrity of the plasma membrane somewhat, resulting in
some antibody being able to enter the cells even when not permeabilized.
The whole blood versus PRP analysis showed that hybridization in whole blood
indicated a greater difference in fluorescence among the genotypes than hybridization in PRP.
It may have been the case that isolating the PRP caused significant enough cell loss that
fluorescence was negatively affected. In addition, centrifuging fixed and permeabilized cells
could have caused some lysis and protein loss, also resulting in lower fluorescence.
Paraformaldehyde at a concentration of 1% was determined to be the optimal
concentration of fixative; Triton at a concentration of 0.5% was determined to provide the best
resolution for detection of protein levels. Paraformaldehyde (1%) was sufficient in fixing the cells
long-term while still being low enough to minimize antigen masking via methylene bridges.
There is a marked decrease in fluorescence seen for fixation with 4% paraformaldehyde as well
as when Saponin was used as a detergent instead of Triton (Figure 4). Saponin was gentler, but
perhaps was not strong enough to permeabilize the cells to the extent that Triton did. Triton was
thus selected to maximize fluorescence, even possibly at the cost of some platelet structural
integrity. Triton at a concentration of 0.1% was sufficient to produce reasonable fluorescence
(Figures 5 and 6) and was selected over 0.5% Triton to minimize damage to platelet
The platelet wash was shown to improve resolution, but not to the extent that it was
required to obtain discernable results from the assay. In the interest of time this step was not
performed in the latter assays hybridizing with three antibodies (Figure 7), but it was utilized in
the assays performed to obtain the data seen in Figure 8.
The triple hybridization indicated a significant difference between intracellular
CalDAG-GEFI in wild-type, heterozygote, and knockout mice. There was some resolution between
intracellular RASA3 in wild-type mice versus heterozygote and knockout mice, but the difference
in expression in heterozygote mice and knockout mice was not as discernable using the Thermo
Fischer antibody. Once the Abcam RASA3 antibody was used instead, resolution improved
greatly, with significant differences being seen between every genotype (Figure 8). The flow
cytometric data also matched up well with the Western blot data, indicating accuracy as well as
precision. In fact, the flow cytometric data for CalDAG-GEFI reflected expected fluorescence
values more accurately than the Western blot data.
This study was the first step towards developing a clinically relevant screening tool for
determining relative expression of RASA3 and CalDAG-GEFI in patients, possibly allowing for
the development of an assay that could be used in a medical setting to evaluate a patient’s
relative risk of thrombocytopenia/thrombosis. Our novel flow cytometry-based assay faithfully
detects RASA3 and CalDAG-GEFI expression in murine platelets, and in future studies we will
use this assay to test whether altered expression of these critical RAP1 regulators is a cause for
hyper- or hypo-reactive platelets in mice and humans.
Acknowledgements
I would like to thank all of the members of the Bergmeier Lab for their generosity and
assistance with many parts of this project. I would also like to specifically acknowledge Dr.
Wolfgang Bergmeier, for providing direction and guidance to my research, and Dr. David Paul,
Works Cited
1. Broos K, Feys HB, Meyer SFD, Vanhoorelbeke K, Deckmyn H. 2011. Platelets at work in
primary hemostasis. Blood Reviews 25:155–167.
2. Crittenden JR, Bergmeier W, Zhang Y, Piffath CL, Liang Y, Wagner DD, Housman DE,
Graybiel AM. 2004. CalDAG-GEFI integrates signaling for platelet aggregation and thrombus
formation. Nature Medicine 10:982–986.
3. Stefanini L, Bergmeier W. 2015. RAP1-GTPase signaling and platelet function. Journal of
Molecular Medicine J Mol Med 94:13–19.
4. Stefanini L, Paul DS, Robledo RF, Chan ER, Getz TM, Campbell RA, Kechele DO, Casari C,
Piatt R, Caron KM, et al. 2015. RASA3 is a critical inhibitor of RAP1-dependent platelet
activation. Journal of Clinical Investigation 125:1419–1432.
5. Stefanini L, Ye F, Snider AK, Sarabakhsh K, Piatt R, Paul DS, Bergmeier W, Petrich BG.
2014. A talin mutant that impairs talin-integrin binding in platelets decelerates IIb 3 activation
without pathological bleeding. Blood 123:2722–2731.
6. Stolla M, Stefanini L, Andre P, Ouellette TD, Reilly MP, Mckenzie SE, Bergmeier W. 2011.
CalDAG-GEFI deficiency protects mice in a novel model of Fc RIIA-mediated thrombosis and