Contents lists available at www.innovativejournal.in
Asian Journal of Computer Science And Information Technology
Journal Homepage: http://innovativejournal.in/ajcsit/index.php/ajcsit
APPLYING DISTRIBUTED PROCESSING FOR DIFFERENT DISTANCE BASED
METHODS DURING PHYLOGENETIC TREE CONSTRUCTION
Pankaj Bhanbri1 , O.P. Gupta2
1Ph.D. Research Scholar,Department of Computer Science and Engineering, I.K.G. Punjab Technical University, Kapurthala, Punjab, India.
2Associate Professor and Head, School of Elect. Engineering and Information Technology, Punjab Agriculture University, Ludhiana, Punjab, India.
ARTICLE INFO ABSTRACT
Corresponding Author:
Pankaj BhanbriPh.D. Research Scholar,
Department of Computer Science and Engineering, I.K.G. Punjab Technical University, Kapurthala, Punjab, India.
Key Words: Phylogenetic tree, Human Protein Reference Database, Human Protein Function, Online Analytical Processing, Support Vector Machines.
DOI:
http://dx.doi.org/10.15520/ajcsit.v7i3.62
For the pharmaceutical industry, the discovery of a new drug presents an enormous scientific challenge, and consists essentially in the identification of the target responsible for the disease. Once the therapeutic target is identified, scientists then find one or more leads that interact with the therapeutic target. Usually leads are searched by employing a long and costly process of trial and error. But if the protein class of the target would have been known it will become very easy to find the complementary lead for the responsible molecule. First data related to human protein is accessed from Human Protein Reference Database (HPRD). From HPRD, the sequences related to ten molecular classes are obtained. For each of the molecular class five amino acid sequences are obtained. Then with the help of various web based tools SDFs are extracted for each sequence. By analyzing the variation in the values of the obtained SDFs priorities are assigned to them. In the present work, tool to neutralize the drug is predicted with the help of distributed processing, which assists to fasten the process as compared to the previous tools used. The tool presented with the help of distributed processing to neutralize the drug ensures a large scope for practical application of the Human Protein Prediction. So, class prediction of a protein supports to shorten the process of drug identification. The simulated results are satisfactory and give the identification of applicability.
©2017, AJCSIT, All Right Reserved. I. INTRODUCTION
Distributed system is a collection of individual computing devices that can communicate with each other. Distributed computing is often used to refer to the implementation of applications on the distributed memory architectures. In the biomedical research field, it is the most widely used form of parallel processing. Parallel processing describes a computing environment where multiple processors cooperate to solve a given computational problem. Distributed processing means the distribution of applications and business logic across multiple processing platforms. Distributed processing implies that the processing will occur on more than one processor in order for a transaction to be completed. In other words, processing is distributed across two or more machines and the processes are most likely not running at the same time, i.e. each process performs part of an application in a sequence. Often the data used in a distributed processing environment is also distributed across platforms. Parallel distributed processing is a model of knowledge representation and information processing according to which items of knowledge are represented by patterns of connections of varying strengths between locations within a network model,
information processing taking the form of parallel processing of collections of activated connections. Distributed Computing refers to the means by which a single computer program runs in more than one computer at the same time. In particular, the different elements and objects of a program are being run or processed using different computer processors. Distributed computing is similar to parallel computing and grid computing. Parallel computing, though, refers to running a single program using a minimum of two processors that belong to one computer. Grid computing, on the other hand, refers to a more dedicated distributed computing setup - one whose computer 'members' are especially dedicated to the program being processed (Hagit Attiya., Jennifer Welch, 2008).
In the simplest sense, parallel computing is the simultaneous use of multiple compute resources to solve a computational problem to be run using multiple CPUs. A problem is broken into discrete parts that can be solved concurrently. Each part is further broken down to a series of instructions. Instructions from each part execute simultaneously on different CPUs.
As proteins are responsible for many different functions in the living cell, it is possible to classify proteins on the basis of their functions as given below:
a. Enzymes: proteins that catalyze chemical and biochemical reactions within living cell and outside. b. Hormones: proteins those are responsible for the regulation of many processes in organisms.
c. Transport proteins: These proteins are transporting or
store some other chemical compounds and ions. d. Antibodies: Proteins that involved into immune
response of the organism to neutralize large foreign molecules, which can be a part of an infection.
e. Structural proteins: These proteins are responsible to maintain structures of other biological components, like cells and tissues.
f. Motor proteins: These proteins can convert chemical energy into mechanical energy.
g. Receptors: These proteins are responsible for signal detection and translation into other type of signal.
h. Signaling proteins: This group of proteins is involved into signaling translation process.
i. Storage proteins: These proteins contain energy, which can be released during metabolism processes in the organism.
III. IMPORTANCE OF PROTEIN CLASSIFICATION The proteins can be divided into various classes on the basis of their functionality. Their importance lies in the process of drug development. An understanding of the classes of proteins is an important component of drug development because proteins are the most common drug targets. Drug development has two major components (Krane and Raymer, 2006):
i) Discovery and ii) Testing
The testing process involves preclinical and clinical trials. The computational methods are not generally subjected to produce significant enhancement in testing processes of drugs. But in the discovery process the computational methods are very helpful. The drug discovery process is labor intensive and expensive and has provided a fertile ground for bioinformatics research. Bioinformatics promises to reduce the labor associated with this process, allowing drugs to be developed faster and at a lower cost.
The drug discovery process itself can be broken into several components including:
a) Target Identification
b) Lead Discovery and Optimization c) Toxicology
d) Pharmacokinetics
a. Target Identification: involves the identification of the target on which the drug acts.
b. Lead Discovery: involves the docking algorithms i.e. the algorithms that help in determining the lead compounds. A lead compound is the compound in the drug which will bind to the target. The optimization includes the use of the database indexing techniques in the docking algorithm to reduce the number of lead compounds by ruling out those that are highly unlikely to bind to the target.
c. Toxicology: involves the study of all the biochemical reactions that will take place when drug is taken.
d. Pharmacokinetics: includes the study of the kinetics of the biochemical reactions.
A disease-causing agent is called pathogen. Target identification involves the identification of a biological molecule that is essential for the survival or proliferation (Multiplication, growth) of the pathogen. Once a target is identified, the objective of the drug design is to develop a molecule that will bind to and inhibit the drug target. Since the functioning of the target is essential to the life processes of the pathogen, inhibition of the target either stops the proliferation of the pathogen or destroys it. An understanding of the structure and function of proteins is an important component of drug development because proteins are the most common drug targets. For example: Human Immunodeficiency Virus (HIV) is a pathogen i.e. it is a disease-causing agent that causes the disease AIDS (Acquired Immune Deficiency Syndrome) (Krane and Raymer, 2002). HIV produces a protein particularly an enzyme called HIV protease, which is essential for the proliferation of the virus HIV or we can say that HIV protease is the drug target. If HIV protease can be effectively inhibited, then the virus cannot affect any additional cells and the advance of the disease can be stopped. After this target identification, the objective of drug design is to discover a molecule that will bind to the HIV protease in a way that prevents it from functioning normally. This is the next step of drug discovery process i.e. lead discovery. The site to which a lead compound can bind to the drug target is extremely specific and is called the active site. Thus, protein class prediction is necessary for the drug discovery process.
The drug discovery process is time consuming and expensive. The process of drug discovery, involves the prediction of protein class based upon existing facts. Sophisticated mining models are needed for protein class prediction. Bioinformatics promises to reduce the labour, time as well as cost associated with this process (Wadhwa, 2006).
IV. BIOINFROMATICS TOOLS FOR SDFs
Following are the various bioinformatics Tools for obtaining sequence derived features (SDFs’):
a. NetNGlyc 1.0 server predicts N-Glycosylation sites in human proteins using artificial neural networks that examine the sequence context of Asn-Xaa-ser/Thr sequins.
b.PSORT server is a computer program for the prediction of protein localization sites in cells. It receives the information of an amino acid sequence and its source origin, e.g., Gram-negative bacteria, as inputs. Then, it analyzes the input sequence by applying the stored rules for various sequence features of known protein sorting signals. Finally, it reports the possibility for the input protein to be localized at each candidate site with additional information.
c.TMHMM server is a program for predicting transmembrane helices based on a hidden Markov model. It reads a FASTA formatted protein sequence and predicts locations of transmembrane, intracellular and extra cellular regions.
d.NetOGlyc server produces neural network predictions of mucin type GalNAc O-Glycosylation sites in mammalian proteins.
signal peptide/non-signal peptide prediction based on a combination of several artificial neural networks and hidden Markov models.
f. Expasy server computes the various physical and chemical parameters for a given protein sequence. The computed parameters include the molecular weight, theoretical pI, amino acid composition, atomic composition, extinction coefficient, estimated half-life, instability index, aliphatic index and grand average of hydropathicity (GRAVY).
g. NetPhos 2.0 server produces neural network predictions for serine, threonine and tyrosine phosphorylation sites in eukaryotic proteins.
V. METHODOLOGY
Class prediction of a protein facilitates to enhance the process of drug discovery (Sita, 2008). In drug discovery, it is very problematic to find out the complementary protein for each protein individually. But if the class of the protein will be known for which the drug is to be discovered then it will become very smooth to find the complementary protein sequence which can be attached to the active site of the protein to stop it to expand. The main concern of the proposed work is the prediction of such a tool to nullify the drug with the help of distributed processing, which assists to fasten the process as compared to the previous tools used. The tool presented with the help of Distributed Processing to neutralize the drug ensures a large scope for practical application of the Human Protein Prediction. So, class prediction of a protein supports to shorten the process of drug identification. The modified technique for protein function prediction will be developed based on the limitations in present techniques.
The methodology of the proposed work includes the prediction of tool to neutralize the drug with the help of distributed processing and study of various data mining algorithms to analyze the working and complexity of diverse algorithms. The predicted tool will be chosen which will provide more accurate predictions to the drug discoverers along with the facility of easy interpretability of learned classification rules as well as helps to fasten the process. Sequence derived features will be utilized to predict the class of protein. The drug discoverer provides unknown protein sequence based on which he wants to predict the class of protein. SDFs can be derived by using web-based tools. These SDFs are then processed into a suitable format that can be utilized to predict class. The model will be tested based on the available classes and existing protein sequences. The data related to human protein is accessed from Human Protein Reference Database (HPRD). From HPRD 5 protein functions are considered and 5 amino acid sequences are extracted for each protein function. These amino acid sequences are then given as input to various web based bioinformatics tools which further provide sequence derived features as output (Surinder, 2008). It includes approximately 163 classes of protein functions. From HPRD, the sequences related to ten molecular classes are obtained. These classes are: Defensin (Def), Heat Shock Protein (HSP), Voltage Gated Channel (VGC), Cell Surface Receptor (CSR), DNA Repair Protein (DRP), Amino peptidase (Ami), Decarboxylase (Dec), G-Protein (GP), RNA Binding Protein (RBP) and Transport/Cargo Protein (T/CP). For each of the molecular class five amino acid sequences are obtained.
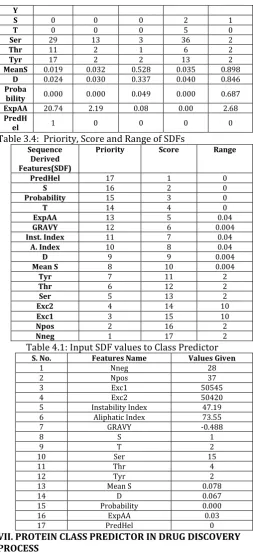
The various bioinformatics tools and their respective SDFs are shown in table 3.1. Values of the SDFs for each of the protein class are stored in the tables 3.2-3.3. Data mining algorithm for Protein Class Prediction is examined. After implementing the algorithm, its complexity is studied and the points are detected, where the distributed processing has to be applied. Now, centering on destination areas, distributed processing is served. Then the distributed processing is embellished in target areas of the data mining algorithm. For transmutation of the algorithm in parallel mode, the algorithm needs to be divided up in labs. Simulation of distributed processing in data mining algorithm is achieved in single system. For the same data mining algorithm, this is carried out in three systems by using cluster. The modified algorithm for Protein Class Predictor using Distributed Environment exemplifies the prediction of protein class by setting number of sequence, number of SDF, credit of sequence, score of SDF and range of SDF with the help of counters in distributed environment. For allocating score to each of the SDF, the variation in the values of each of the SDFs is studied for each functional class. High priority features are the large variation of the value of particular SDF from class to class. Low priority features are the less variation of the value of SDF from class to class. On the basis of priority, score is allocated to each SDF. Priority and score allocated to each of the SDF is shown in Table 3.4. This score is utilized to find the highest score sequence to predict the class of the entered sequence.
Range of the SDFs is used to determine the similarity between entered values of the SDFs and the sequences stored into the database. For allocating range to each of the SDF, the difference in the values of each of the SDFs is analyzed for each sequence and functional class. Values of four lowest priority features, PredHel, S, T and Probability are repeated heavily from sequence to sequence and from class to class, so these features are supposed to match exactly to find the similar sequences, so no range is allocated to them. For the remaining features, differences in the values are studied thoroughly for each and range is allocated to each feature so that it should give rise to least overlap in the values from sequence to sequence or from class to class.
VI. ALGORITHM FOR PROTEIN CLASS PREDICTOR a. The cluster algorithm for protein class predictor is
implemented for seventeen sequence derived features and ten protein classes. Five amino acid sequences are considered in each class. The computation is enhanced by identifying the different areas in the code where the distributed processing can be implemented. Then the distributed processing is embedded in those areas having four worker labs.
b. The modified algorithm for Protein Class Predictor using Distributed Environment has been shown as: Step 1: Start Algorithm
Step 2: Enter features Fj Step 3: Set i = 1, j =1, Ci =0
Step 4: Check the value of Aij Step 5: Check whether Fj+Rj/2>=Aij>=Fj-Rj/2, If yes then
Ci=Ci+Sj, otherwise check the value of i and repeat step 4 to step 5.
previous cluster to ith cluster and set Ci=Ci+Sj otherwise i=i+1, j=j+1 and and repeat step 5 to step 6.
(Distributed Processing is applied in this loop by
distributing the loop computation in four labs)
Step 7: Check If i=M, if no, then i=i+1 otherwise check j=N, if no, then j=j+1 otherwise set K=N (Highest Priority Cluster),Maxc=0, Maxp=0.
(Distributed Processing is applied in this loop by
distributing the loop computation in four labs)
Step 8: Set Maxc = MaxK (Maximum credit in kth cluster). Step 9: Check If Maxc=MpK, If yes, then display the Maxc sequence class, otherwise check the value of K. If K=1 (Lowest priority cluster), then display the Maxc sequence class, otherwise set K = K-1 and Maxp=Maxc and repeat step 8 to step 9.
(Distributed Processing is applied in this loop by
distributing the loop computation in four labs) Step 10: End Algorithm.
Table 3.1: SDFs’ Obtained from Web Based Tools Tool used SDFs’ Obtained
ExPASy ProtParam Extinction Coefficients
No. of negatively charged residues No. of positively charged residues Instability Index
Aliphatic Index GRAVY
NetOGlyc O-Glycosylation sites
NetPhos Sr and Thr phosphorylation
Tyr phosphorylation
SignalP Signal Peptide
TMHMM Transmembrane Helices
Table 3.2: Values of SDFs for Protein Classes
SDFs Protein Class
(Amino Acid Sequences)
Defensi n (94AA) Heat Shock (573AA) Voltage Gated Channel (1091AA) Cell Surface Recepto r (556AA) DNA Repair Protein (318AA)
Nneg 9 80 142 73 43
Npos 9 75 114 49 46
Exc1 15845 16055 166270 114860 56755
Exc2 15470 15930 165020 114360 56380
I.Index 49.71 30.19 34.74 59.09 43.41
A.Index 102.02 100.73 84.97 68.58 77.36
GRAVY 0.285 -0.076 -0.345 -0.576 -0.583
S 3 2 0 2 1
T 0 0 0 0 0
Ser 0 12 24 29 6
Thr 7 7 11 5 2
Tyr 0 5 18 10 4
MeanS 0.579 0.436 0.850 0.848 0.062
D 0.353 0.334 0.646 0.784 0.041
Probabi
lity 0.181 0.147 0.323 0.992 0.000
ExpAA 0.64 0.01 11.04 24.79 0.04
PredHel 0 0 0 1 0
Table 3.3: Values of SDFs for Protein Classes
SDFs Protein Class
(Amino Acid Sequences) Amino-peptida se (1025A A) Decarbo xylase (480AA) G-Protein (180AA) RNA-Binding Protein (526AA) Transpo rt/Cargo Protein (189AA)
Nneg 122 55 22 37 20
Npos 97 53 21 51 15
Exc1 178580 69620 29575 70390 32680
Exc2 178080 68870 29450 70140 32430
I.Inde
x 40.94 40.13 46.47 57.11 40.92
A.Inde
x 89.02 89.27 94.72 13.38 95.45
GRAV -0.194 -0.040 -0.204 -1.319 -0.053
Y
S 0 0 0 2 1
T 0 0 0 5 0
Ser 29 13 3 36 2
Thr 11 2 1 6 2
Tyr 17 2 2 13 2
MeanS 0.019 0.032 0.528 0.035 0.898
D 0.024 0.030 0.337 0.040 0.846
Proba
bility 0.000 0.000 0.049 0.000 0.687
ExpAA 20.74 2.19 0.08 0.00 2.68
PredH
el 1 0 0 0 0
Table 3.4: Priority, Score and Range of SDFs Sequence
Derived Features(SDF)
Priority Score Range
PredHel 17 1 0
S 16 2 0
Probability 15 3 0
T 14 4 0
ExpAA 13 5 0.04
GRAVY 12 6 0.004
Inst. Index 11 7 0.04
A. Index 10 8 0.04
D 9 9 0.004
Mean S 8 10 0.004
Tyr 7 11 2
Thr 6 12 2
Ser 5 13 2
Exc2 4 14 10
Exc1 3 15 10
Npos 2 16 2
Nneg 1 17 2
Table 4.1: Input SDF values to Class Predictor
S. No. Features Name Values Given
1 Nneg 28
2 Npos 37
3 Exc1 50545
4 Exc2 50420
5 Instability Index 47.19
6 Aliphatic Index 73.55
7 GRAVY -0.488
8 S 1
9 T 2
10 Ser 15
11 Thr 4
12 Tyr 2
13 Mean S 0.078
14 D 0.067
15 Probability 0.000
16 ExpAA 0.03
17 PredHel 0
VII. PROTEIN CLASS PREDICTOR IN DRUG DISCOVERY PROCESS
the value of a sequence will be in the specified range of the entered value then that sequence will be included in the cluster of that feature. And every time a sequence will enter a cluster its credit will be incremented by the score of that feature. So in this manner clusters for all the SDFs will be generated.
After all the clusters will be generated they will be backtracked to find the highest credit sequence. Starting with the highest priority cluster, sequence with the maximum credit will be determined from the current cluster. If the maximum credit obtained from the current cluster will be greater than the previous cluster then new sequence will become the maximum credit sequence other wise previous sequence will remain the highest credit sequence. While traveling from higher priority clusters to lower priority, in each of the cluster if the maximum score of the current credit will be greater than all the previous clusters and also equal to the maximum possible credit of that cluster then the sequence with this credit will be the maximum credit sequence in the whole database and need not to travel further. Then the class of the sequence with maximum credit will be included in the prediction result and the result will be Heat Shock Protein. The tool presented with the help of Distributed Processing to neutralize the drug ensures a large scope for practical application of the Human Protein Prediction. So, class prediction of a protein supports to shorten the process of drug identification. Data mining algorithm for Protein Class Prediction is examined. After implementing the algorithm, its complexity is studied and the points are located, where the distributed processing has to be applied. Now, centering on destination areas, distributed processing is served. Then the distributed processing is embellished in target areas of the data mining algorithm. For transmutation of the algorithm in parallel mode, the algorithm needs to be divided up in labs. Simulation of distributed processing in data mining algorithm is achieved in single system with the help of plotting the timing graph ( cpu time and real time). For the same data mining algorithm, this is carried out in three systems by using cluster. The tool presented with the help of Distributed Processing to neutralize the drug ensures a large scope for practical application of the Human Protein Prediction.
VIII. CONCLUSION AND FUTURE SCOPE
This work is designed and implemented for prediction of tool to neutralize the drug with the help of distributed processing and study of various data mining algorithms to analyze the working and complexity of diverse algorithms. The predicted tool is chosen, which provides more accurate predictions to the drug discoverers along with the facility of easy interpretability of learned classification rules as well as helps to fasten the process. Sequence derived features are utilized to predict the class of protein. The drug discoverer provides unknown protein sequence based on which he wants to predict the class of protein. SDFs can be derived by using web-based tools. These SDFs are then processed into a suitable format that can be utilized to predict class.
The modified algorithm for Protein Class Predictor using Distributed Environment exemplifies the prediction of protein class by setting number of sequence, number of SDF, credit of sequence, score of SDF and range of SDF with the help of counters in distributed environment. Data mining algorithm for Protein Class
Prediction is examined. After implementing the algorithm, its complexity is studied and the points are detected, where the distributed processing has to be applied. Now, centering on destination areas, distributed processing is served. Then the distributed processing is embellished in target areas of the data mining algorithm. For transmutation of the algorithm in parallel mode, the algorithm needs to be divided up in labs. Simulation of distributed processing in data mining algorithm is achieved in single system with the help of plotting the timing graph ( cpu time and real time). By examining the cpu time and real time of parfor and batch, parfor takes lesser cpu and real time as compared to batch and hence gives the better result. For the same data mining algorithm, this is carried out in three systems by using cluster. The tool presented with the help of Distributed Processing to neutralize the drug ensures a large scope for practical application of the Human Protein Prediction. Following is the scope for further work in this class predictor model:
Different prokaryotes can be added for the fast and efficient implementation of Distributed Processing. Automatic generation of Sequence derived features can
be done.
More Sequence derived features can be explored. REFERENCES
1. Bajorath, J., Klein, T.E., Lybrand, T.P. and Novotony, J. (1999) “Computer –Aided Drug Discovery: From Target Proteins to Drug Candidates”, Pacific Symposium on Bio Computing, vol. 4, pp. 413-414.
2. Bartoli, S., Fincham, C.I. and Fatorri, D. (2006) “The
fragment-approach: An update”, Drug Discovery
Today: Technologies, vol. 3, issue 4, pp. 425-431. 3. Bergeron, B. (2003) “Bioinformatics Computing”,
Pearson Education, pp. 110- 160.
4. Brune, C., Chevenet, F., Martin, D., Wojcik, J., Guenoche, A. and Jacq, B. (2003) “Functional Classification of Proteins for the Prediction of Cellular Function from a
Protein – Protein Interaction Network”, Genome
Biology, vol. 5, pp. 6-13.
5. Devos, D. and Valencia, A. (2000) “Practical Limits of Function Prediction”, Protein Design Group, National Centre for Biotechnology, CNB-CSICMadrid, E-28049, Spain, pp. 134-170.
6. Huan, J., Wang, W., Washington, A., Prins, J., Shah, R. and Tropsha, A. (2004), “Accurate Classification of Protein Structural Families Using Coherent Sub graph
Analysis”, in Proceedings of the Pacific Symposium on
Biocomputing (PSB),pp. 411-422.
7. Jacobson, M. and Sali, A. (2004) “Comparative Protein Structure Modelling and its Applications to Drug Discovery”, Annual Reports in medicinal Chemistry, vol. 39,pp.259-273.
8. Jensen, L., Skovgaard, M. and Brunak, S. (2002) “Prediction of Novel Archaea Enzymes from Sequence
Derived Features”, Protein Science, Technical
University Of Denmark, vol. 11, pp. 2894-2898. 9. Jensen, L. (2002) “Prediction of Protein Function from
Sequence Derived Protein Features”, Ph.D. thesis, Technical University of Denmark, pp. 1-570.
10. Kang, L., Chung, B.G., Langer, R. and Khademhosseini (2008) “Microfluidics for drug discovery and development: From target selection to product lifecycle
management”, Drug Discovery Today, vol. 13, issue
and Wagg, J. (1999) “Integrated pathway/ genome
databases and their role in Drug Discovery”, Trends in
Biotechnology, vol.17,issue 7, pp. 275-281.
11. Karwath, A. and Raedt, L. (2004) “Predictive Graph
Mining” Springer – Verlag Berlin Heidelberg, LNAI
3245, pp. 1-15.
12. Kaur, S. (2008) “Decision Tree Classifier for Human Protein Function Prediction”, M.Tech. Thesis, Punjab Technical University, pp. 13-34.
13. Krane, D. and Raymer, M. (2006) “Fundamental
Concepts of Bioinformatics”, Pearson Education
Publishers.
14. Laxman, S. and Sastry, P. S. (2006) “A Survey of Temporal Data Mining” Sadhana, vol. 32, Part 2, April, 2006, pp. 173-198.
15. Mark, B. and Brocklebank, J. (2005) “Data Mining”, SAS Institute Inc Data Mining, vol.6, pp 123-127.
16. Merschmann, Luiz and Plastino, Alexandre (2007) “A
Lazy Data Mining Approach for Protein Classification”,
Nanobioscience, vol.6, issue 1, March, 2007, pp. 36-42. 17. Park, C.Y., Park, S.H., Kim, D.H., Park, S.H. and Hwang,
C.J. (2004) “A new protein Classification method using dynamic classifier”, Bioinformatics, vol. 9, pp 32-35. 18. Psomopoulos, F. E. and Mitkas, P. A. (2005), “A protein
classification engine based On stochastic finite state
automata”, in proceedings of Symposium 35:
Computational Methods in Molecular Biology at the ICCMSE 2005, Loutraki, Greece, October, 2005, pp. 1371-1374.
19. Rastogi, S. C., Mendiratta, N. and Rastogi, P. (2005)
“Bioinformatics Methods and Applications”, third
edition, PHI publication, pp.1-350.
20. Rani, S. (2008) “Protein Class Prediction using Graph Mining”, M.Tech. Thesis, Punjab Technical University, pp. 13-34.
21. Samola, A. and Schonauer, S. (2004) “Graph Based Functional Classifications of Proteins Using Kernel
Method”, Karsten Borgwardt, Chapter 2, pp. 28-65.
22. Sahoo, S.K., Dilnawaz, F. and Krishnakumar, S. (2008)
“Nanotechnology in ocular drug delivery”, Drug
Discovery Today, vol. 13, issue 3-4, pp. 144-151. 23. Singh, M., Singh, P. and Singh, H. (2006), “Decision
Tree Classifier for Human Protein Function Prediction”,
in Proceedings of International Conference on Advanced Computing and Communication ADCOM 2006, December, 2006, pp. 564-568.
24. Singh M., Wadhwa P. and Sandhu P. (2007) “Human Protien Function Prediction using Decision Tree induction” IJCSNS vol.7 no.4, pp 92-98.
25. Veenstra, T.D. (2006) “Proteomic approaches in drug discovery”, Drug Discovery Today: Technologiese, vol. 3, issue 4, pp. 433-440.
26. Wadhwa, P.K. (2006), “Development of Data Mining
Model for Human Protein Function Prediction”, M-Tech
Thesis, Punjab Technical University, pp. 1-40.
27. Xiao-Li, L., Soon-Hang, T., Chuan-Sheng, F. and See-Kiong, N. (2005) “Interaction Graph Mining for Protein
Complexes Using Local Clique Merging”, Genome
Informatics, vol. 16(2), pp. 260-269.
A.1 Screen Shots
a. Protein Class Predictor using parfor and matlabpool in real time
c. Protein Class Predictor using batch and matlabpool in real time
f. Protein Class Predictor using Profiler
g. Distributed Processing in Protein Class Predictor