Acta Cryst.(2003). E59, o1073±o1075 DOI: 10.1107/S1600536803014041 K. Umadeviet al. C4H7NO4H2O
o1073
organic papers
Acta Crystallographica Section E Structure Reports
Online
ISSN 1600-5368
L
-Aspartic acid monohydrate
K. Umadevi,aK. Anitha,a B. Sridhar,aN. Srinivasanb and R. K. Rajarama*
aDepartment of Physics, Madurai Kamaraj
University, Madurai 625 021, India, and
bDepartment of Physics, Thiagarajar College,
Madurai 625 009, India
Correspondence e-mail: [email protected]
Key indicators
Single-crystal X-ray study
T= 293 K
Mean(C±C) = 0.004 AÊ
Rfactor = 0.061
wRfactor = 0.173
Data-to-parameter ratio = 12.2
For details of how these key indicators were automatically derived from the article, see http://journals.iucr.org/e.
#2003 International Union of Crystallography Printed in Great Britain ± all rights reserved
In the title compound, C4H7NO4H2O, the screw-related
aspartic acid molecules are linked along the a axis by NÐ
H O hydrogen bonds to form helical structures. The
adjacent helices are inter-linked through OÐH O hydrogen
bonds and also by the water molecules through NÐH O(W)
and O(W)ÐH O hydrogen bonds, to form a
three-dimen-sional network.
Comment
Aspartic acid is a non-essential amino acid, widely distributed in proteins, which plays a major role in the energy cycle of the
human body. The crystal structures of l-aspartic acid
(Derissenet al., 1968),dl-aspartic acid (Rao, 1973; Sequeiraet
al., 1989), dl-aspartic acid nitrate monohydrate (Asath
Bahadur & Rajaram, 1995), bis(dl-aspartic acid) sulfate
(Srinivasanet al., 2001) andl-aspartic acid nitrate±l-aspartic
acid (1/1) (Sridhar et al., 2002) have been reported. In the
present paper, the crystal structure ofl-aspartic acid
mono-hydrate, (I), is reported.
The asymmetric unit of (I) contains one aspartic acid molecule and one water molecule (Fig. 1). The equality of CÐ O bond distances [1.240 (4) and 1.259 (4) AÊ] and OÐCÐC
bond angles [118.3 (3) and 115.3 (3)] (Table 1) characterize
the deprotonated carboxylate group. The backbone
conformation angle 1 of ÿ10.8 (4) indicates the cisform.
The side chain shows a gauche II conformation [1 =
ÿ71.8 (3)]. The branched chain conformation angles11and
21correspond to thecisandtransforms. The Catom is in the gaucheI [52.7 (3)] conformation with respect to the C0atom.
The molecular structure is stabilized by a weak intramolecular
N1ÐH1B O3 hydrogen bond.
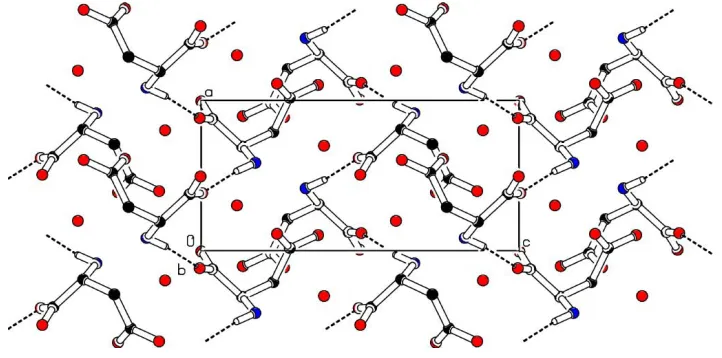
The screw-related aspartic acid molecules are linked along
the a axis by N1ÐH1A O1i hydrogen bonds to form a
helical structure (Table 2 and Fig. 2). This helical structure is
further stabilized by N1ÐH1C O3iihydrogen bonds, which
link the molecules translated by one unit along theaaxis. The
adjacent helices are interlinked through O4ÐH4 O2iii
hydrogen bonds and also by the water molecules through
N1ÐH1B O11iv, O11ÐH11 O2v and O11ÐH12 O2i
organic papers
o1074
K. Umadeviet al. C4H7NO4H2O Acta Cryst.(2003). E59, o1073±o1075hydrogen bonds, to form a three-dimensional network. Within
the network, the O4ÐH4 O2iii hydrogen bonds link the
screw-related molecules, to form zigzag chains along thecaxis. A class II hydrogen-bonding pattern is observed in the present structure, having two two-centered hydrogen bonds and one three-centered hydrogen bond (Jeffrey & Saenger, 1991). In the present study, the water molecule shows a planar 1B-1/1D
orientation (Jeffrey & Saenger, 1991). All the symmetry codes
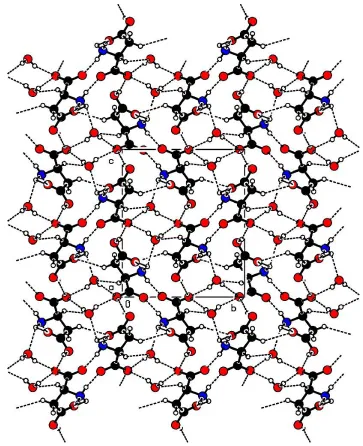
are as in Table 2. A view of the molecular packing down thea
axis is shown in Fig. 3.
Experimental
The title compound was crystallized from an aqueous solution when attempts were made to grow single crystals of a complex ofl-aspartic acid with sulfuric acid.
Crystal data
C4H7NO4H2O Mr= 151.12
Orthorhombic,P212121 a= 5.587 (4) AÊ
b= 9.822 (5) AÊ
c= 11.813 (9) AÊ
V= 648.2 (8) AÊ3 Z= 4
Dx= 1.548 Mg mÿ3
Dm= 1.54 Mg mÿ3
Dmmeasured by ¯otation in a
mixture of carbon tetrachloride and xylene
MoKradiation Cell parameters from 25
re¯ections
= 8.0±13.8 = 0.14 mmÿ1 T= 293 (2) K Block, colorless 0.30.30.3 mm
Data collection
Enraf±Nonius CAD-4 diffractometer
!±2scans
Absorption correction: scan (Northet al., 1968)
Tmin= 0.958,Tmax= 0.958
1318 measured re¯ections 1132 independent re¯ections 1091 re¯ections withI> 2(I)
Rint= 0.085 max= 24.6 h= 0!6
k= 0!11
l=ÿ14!14 3 standard re¯ections
frequency: 60 min intensity decay: none
Re®nement
Re®nement onF2 R[F2> 2(F2)] = 0.061 wR(F2) = 0.173 S= 1.06 1132 re¯ections 93 parameters
H atoms treated by a mixture of independent and constrained re®nement
w= 1/[2(F
o2) + (0.136P)2
+ 0.3381P]
whereP= (Fo2+ 2Fc2)/3
(/)max< 0.001
max= 0.33 e AÊÿ3
min=ÿ0.54 e AÊÿ3
Table 1
Selected geometric parameters (AÊ,).
O1ÐC1 1.240 (4)
O2ÐC1 1.259 (4) C4ÐO3C4ÐO4 1.244 (4)1.322 (4) O1ÐC1ÐO2 126.2 (3)
O1ÐC1ÐC2 118.3 (3) O2ÐC1ÐC2 115.3 (3) O1ÐC1ÐC2ÐN1 ÿ10.8 (4)
O2ÐC1ÐC2ÐN1 173.9 (3) O1ÐC1ÐC2ÐC3 ÿ136.2 (3) O2ÐC1ÐC2ÐC3 48.5 (3)
N1ÐC2ÐC3ÐC4 ÿ71.9 (3) C1ÐC2ÐC3ÐC4 52.8 (3) C2ÐC3ÐC4ÐO3 ÿ2.9 (4) C2ÐC3ÐC4ÐO4 177.0 (3)
Figure 2
A view of the helical structures formed along theaaxis. For clarity, all H
atoms except H1Ahave been omitted.
Figure 3
Hydrogen-bonding network, viewed down theaaxis.
Figure 1
Table 2
Hydrogen-bonding geometry (AÊ,).
DÐH A DÐH H A D A DÐH A
N1ÐH1A O1i 0.89 1.93 2.813 (4) 171
N1ÐH1C O3ii 0.89 2.02 2.809 (4) 147
O4ÐH4 O2iii 0.82 2.15 2.933 (4) 161
N1ÐH1B O11iv 0.89 2.22 2.854 (4) 128
O11ÐH11 O2v 0.78 2.06 2.837 (4) 170
O11ÐH12 O2i 0.80 2.05 2.817 (4) 160
N1ÐH1B O3 0.89 2.55 3.093 (4) 120
Symmetry codes: (i) xÿ12;1
2ÿy;ÿz; (ii) xÿ1;y;z; (iii) 52ÿx;ÿy;12z; (iv) 1ÿx;yÿ12;1
2ÿz; (v)xÿ1;1y;z.
The H atoms of the water molecule were located from a difference Fourier map and their isotropic displacement parameters were re®ned [Uiso(H) = 0.04 (1) and 0.07 (2) AÊ2]. All other H atoms were placed in geometrically calculated positions and included in the re®nement in the riding-model approximation, with Uiso equal to 1.2Ueqof the carrier atom. The data set includes 442 Friedel pairs; however, the lack of any signi®cant anomalous effects precludes the con®rmation of the absolute con®guration from the diffraction data, and it has been assumed.
Data collection: CAD-4 Software (Enraf±Nonius, 1989); cell re®nement: CAD-4 Software; data reduction: CAD-4 Software; program(s) used to solve structure: SHELXS97 (Sheldrick, 1997); program(s) used to re®ne structure:SHELXL97 (Sheldrick, 1997); molecular graphics:PLATON(Spek, 1999); software used to prepare material for publication:SHELXL97.
BS thanks the Council of Scienti®c and Industrial Research (CSIR), Government of India, for ®nancial assistance. RKR thanks the Department of Science and Technology (DST), Government of India, for ®nancial support. Financial support from the UGC is also gratefully acknowledged.
References
Asath Bahadur, S. & Rajaram. R. K. (1995).Z. Kristallogr.210, 276±278. Derissen, J. L., Endeman, H. J. & Peerdeman, A. F. (1968).Acta Cryst.B24,
1349±1354.
Enraf±Nonius (1989).CAD-4Software. Version 5.0. Enraf±Nonius, Delft, The Netherlands.
Jeffrey, G. A. & Saenger, W. (1991). In Hydrogen Bonding in Biological Structures. Berlin, Heidelberg, New York: Springer-Verlag.
Johnson, C. K. (1976).ORTEPII. Report ORNL-5138. Oak Ridge National Laboratory, Tennessee, USA.
North, A. C. T., Phillips, D. C. & Mathews, F. S. (1968).Acta Cryst.A24, 351± 359.
Rao, S. T. (1973).Acta Cryst.B29, 1718±1720.
Sequeira, A., Rajagopal, H. & Ramanadham, M. (1989).Acta Cryst.C45, 906± 908.
Sheldrick, G. M. (1997). SHELXL97 and SHELXS97. University of GoÈttingen, Germany.
Spek, A. L. (1999).PLATON.Utrecht University, The Netherlands. Sridhar, B., Srinivasan, N. & Rajaram, R. K. (2002).Acta Cryst.E58, o1372±
o1374.
Srinivasan, N., Sridhar, B. & Rajaram, R. K. (2001).Acta Cryst.E57, o679± o681.
supporting information
sup-1
Acta Cryst. (2003). E59, o1073–o1075supporting information
Acta Cryst. (2003). E59, o1073–o1075 [doi:10.1107/S1600536803014041]
L
-Aspartic acid monohydrate
K. Umadevi, K. Anitha, B. Sridhar, N. Srinivasan and R. K. Rajaram
S1. Comment
Aspartic acid is non-essential amino acid, widely distributed in proteins, which plays a major role in the energy cycle of
the human body. The crystal structures of L-aspartic acid (Derissen et al., 1968), DL-aspartic acid (Rao, 1973; Sequeria et
al., 1989), DL-aspartic acid nitrate monohydrate (Asath Bahadur & Rajaram, 1995), bis(DL-aspartic acid) sulfate
(Srinivasan et al., 2001) and L-aspartic acid nitrate–L-aspartic acid (1/1) (Sridhar et al., 2002) have been reported. In the
present paper, the crystal structure of L-aspartic acid monohydrate, (I), is reported.
The asymmetric unit of (I) contains one aspartic acid residue and one water molecule (Fig. 1). The equality of C—O
bond distances [1.240 (4) and 1.259 (4) Å] and O—C—C bond angles [118.3 (3) and 115.3 (3)°] (Table 1) represent the
deprotonated carboxylate group. The backbone conformation angle ψ1 of −10.8 (4)° indicates the cis form. The side chain
shows a gauche II conformation [χ1 = −71.8 (3)°]. The branched chain conformation angles χ11 and χ21 are in cis and trans
form. The Cγ atom is in the gauche I [52.7 (3)°] conformation with respect to C′ atom. The molecular structure is
stabilized by a weak intramolecular N1—H1B···O3 hydrogen bond.
The screw-related aspartic acid molecules are linked along the a axis by N1—H1A···O1i hydrogen bonds to form a
helical structure (Table 2 and Fig.1). This helical structure is further stabilized by N1—H1C···O3ii hydrogen bonds which
link the molecules translated a unit along the a axis. The adjacent helices are interlinked through O4—H4···O2iii hydrogen
bonds and also by the water molecules through N1—H1B···O11iv, O11—H11···O2v and O11—H12···O2i hydrogen bonds,
to form a three-dimensional network. Within the network, the O4—H4···O2iii hydrogen bonds link the screw related
molecules, to form zigzag chains along the c axis. Class II hydrogen-bonding pattern is observed in the present structure
having two two-centered hydrogen bonding and one three-centered hydrogen bonding (Jeffrey & Saegner, 1991). In the
present study, the water molecule shows planar 1B-1/one-dimensional orientation (Jeffrey & Saegner, 1991). All the
symmetry codes are as in Table 2. A view of the molecular packing down the a axis is shown in Fig. 2.
S2. Experimental
The title compound was crystallized from the aqueous solution when attempts were made to grow the single crystals of
L-aspartic acid with sulfuric acid.
S3. Refinement
The H atoms of the water molecule were located from a difference Fourier map and their isotropic displacement
parameters were refined [Uiso(H) = 0.04 (1) and 0.07 (2) Å2]. All other H atoms were placed in geometrically calculated
positions and included in the refinement in the riding-model approximation, with Uiso equal to 1.2Ueq of the carrier atom.
Intensities for 442 Friedel pairs were measured, resulting in a Flack parameter of 0(3). Though the absolute structure
could not be confirmed as a result of weak anamalous signal, the Friedel pairs were not merged due to resulting low r/p
supporting information
[image:5.610.125.486.264.443.2]sup-2
Acta Cryst. (2003). E59, o1073–o1075Figure 1
The structure of the title compound, showing 50% probability displacement ellipsoids (Johnson, 1976) and the
atom-numbering scheme.
Figure 2
supporting information
[image:6.610.122.485.69.515.2]sup-3
Acta Cryst. (2003). E59, o1073–o1075Figure 3
Hydrogen-bonding network viewed down the a axis.
′L-aspartic acid monohydrate′
Crystal data
C4H7NO4·H2O
Mr = 151.12
Orthorhombic, P212121 Hall symbol: P 2ac 2ab
a = 5.587 (4) Å
b = 9.822 (5) Å
c = 11.813 (9) Å
V = 648.2 (8) Å3
Z = 4
F(000) = 320
Dx = 1.548 Mg m−3
Dm = 1.54 Mg m−3
Dm measured by Flotation in a mixture of carbon tetrachloride and xylene
Mo Kα radiation, λ = 0.70165 Å Cell parameters from 25 reflections
θ = 8.0–13.8°
µ = 0.14 mm−1
supporting information
sup-4
Acta Cryst. (2003). E59, o1073–o1075Data collection
Enraf-Nonius CAD-4 diffractometer
Radiation source: fine-focus sealed tube Graphite monochromator
ω–2θ scans
Absorption correction: ψ scan (North et al., 1968)
Tmin = 0.958, Tmax = 0.958 1318 measured reflections
1132 independent reflections 1091 reflections with I > 2σ(I)
Rint = 0.085
θmax = 24.6°, θmin = 2.7°
h = 0→6
k = 0→11
l = −14→14
3 standard reflections every 60 min intensity decay: none
Refinement
Refinement on F2 Least-squares matrix: full
R[F2 > 2σ(F2)] = 0.061
wR(F2) = 0.173
S = 1.06 1132 reflections 93 parameters 0 restraints
Primary atom site location: structure-invariant direct methods
Secondary atom site location: difference Fourier map
Hydrogen site location: inferred from neighbouring sites
H atoms treated by a mixture of independent and constrained refinement
w = 1/[σ2(F
o2) + (0.136P)2 + 0.3381P] where P = (Fo2 + 2Fc2)/3
(Δ/σ)max < 0.001 Δρmax = 0.33 e Å−3 Δρmin = −0.54 e Å−3
Special details
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes.
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger.
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
x y z Uiso*/Ueq
supporting information
sup-5
Acta Cryst. (2003). E59, o1073–o1075H4 1.2247 −0.0242 0.3907 0.068* O11 0.3015 (6) 0.7701 (3) 0.1132 (2) 0.0430 (7) H11 0.2037 0.8162 0.0855 0.037 (11)* H12 0.3390 0.7142 0.0674 0.072 (19)*
Atomic displacement parameters (Å2)
U11 U22 U33 U12 U13 U23
O1 0.0359 (11) 0.0217 (11) 0.0268 (11) −0.0021 (9) 0.0069 (10) 0.0077 (10) O2 0.0363 (12) 0.0256 (11) 0.0348 (12) 0.0058 (10) 0.0098 (11) −0.0020 (10) C1 0.0208 (14) 0.0244 (14) 0.0179 (13) −0.0016 (11) 0.0004 (11) 0.0007 (12) C2 0.0206 (14) 0.0162 (14) 0.0246 (15) −0.0016 (12) 0.0014 (12) −0.0001 (11) N1 0.0232 (12) 0.0187 (12) 0.0256 (13) 0.0034 (10) 0.0022 (10) 0.0019 (10) C3 0.0257 (15) 0.0171 (14) 0.0280 (15) −0.0026 (12) 0.0053 (13) 0.0077 (12) C4 0.0220 (13) 0.0180 (14) 0.0244 (14) 0.0025 (11) 0.0054 (12) 0.0027 (12) O3 0.0275 (11) 0.0237 (12) 0.0391 (12) −0.0059 (9) −0.0041 (10) 0.0068 (10) O4 0.0454 (16) 0.0453 (16) 0.0445 (14) −0.0009 (14) −0.0070 (14) 0.0067 (13) O11 0.0516 (16) 0.0395 (14) 0.0380 (14) 0.0107 (13) −0.0034 (13) −0.0022 (12)
Geometric parameters (Å, º)
O1—C1 1.240 (4) C3—C4 1.522 (5) O2—C1 1.259 (4) C3—H3A 0.97 C1—C2 1.538 (4) C3—H3B 0.97 C2—N1 1.499 (3) C4—O3 1.244 (4) C2—C3 1.519 (4) C4—O4 1.322 (4) C2—H2 0.98 O4—H4 0.82 N1—H1A 0.89 O11—H11 0.78 N1—H1B 0.89 O11—H12 0.80 N1—H1C 0.89
O1—C1—O2 126.2 (3) H1A—N1—H1C 109.5 O1—C1—C2 118.3 (3) H1B—N1—H1C 109.5 O2—C1—C2 115.3 (3) C2—C3—C4 113.6 (2) N1—C2—C3 110.9 (2) C2—C3—H3A 108.8 N1—C2—C1 109.5 (2) C4—C3—H3A 108.8 C3—C2—C1 114.6 (2) C2—C3—H3B 108.8 N1—C2—H2 107.2 C4—C3—H3B 108.8 C3—C2—H2 107.2 H3A—C3—H3B 107.7 C1—C2—H2 107.2 O3—C4—O4 123.0 (3) C2—N1—H1A 109.5 O3—C4—C3 120.8 (3) C2—N1—H1B 109.5 O4—C4—C3 116.2 (3) H1A—N1—H1B 109.5 C4—O4—H4 109.5 C2—N1—H1C 109.5 H11—O11—H12 107.5
supporting information
sup-6
Acta Cryst. (2003). E59, o1073–o1075O2—C1—C2—C3 48.5 (3) C2—C3—C4—O4 177.0 (3)
Hydrogen-bond geometry (Å, º)
D—H···A D—H H···A D···A D—H···A
N1—H1A···O1i 0.89 1.93 2.813 (4) 171 N1—H1C···O3ii 0.89 2.02 2.809 (4) 147 O4—H4···O2iii 0.82 2.15 2.933 (4) 161 N1—H1B···O11iv 0.89 2.22 2.854 (4) 128 O11—H11···O2v 0.78 2.06 2.837 (4) 170 O11—H12···O2i 0.80 2.05 2.817 (4) 160 N1—H1B···O3 0.89 2.55 3.093 (4) 120