organic papers
o2178
Funet al. C11H10ClN3O2S doi:10.1107/S1600536806015832 Acta Cryst.(2006). E62, o2178–o2180
Acta Crystallographica Section E
Structure Reports
Online
ISSN 1600-5368
N-(6-Chloropyridazin-3-yl)-4-methylbenzene-sulfonamide
Hoong-Kun Fun,a* Shu-Feng Zhang,bZhen-Feng Chen,b Hong Liangband
Suchada Chantraprommac*
aX-ray Crystallography Unit, School of Physics,
Universiti Sains Malaysia, 11800 USM, Penang, Malaysia,bSchool of Chemistry and Chemical Engineering, Guangxi Normal University, Guilin 541004, People’s Republic of China, and cDepartment of Chemistry, Faculty of Science,
Prince of Songkla University, Hat-Yai, Songkhla 90112, Thailand
Correspondence e-mail: hkfun@usm.my, suchada.c@psu.ac.th
Key indicators
Single-crystal X-ray study T= 297 K
Mean(C–C) = 0.003 A˚ Rfactor = 0.050 wRfactor = 0.159
Data-to-parameter ratio = 20.9
For details of how these key indicators were automatically derived from the article, see http://journals.iucr.org/e.
Received 27 April 2006 Accepted 28 April 2006
#2006 International Union of Crystallography All rights reserved
In the title compound, C11H10ClN3O2S, the pyridazine ring
and the benzene ring adopt a distorted V configuration, forming a dihedral angle of 73.79 (11). The crystal packing is
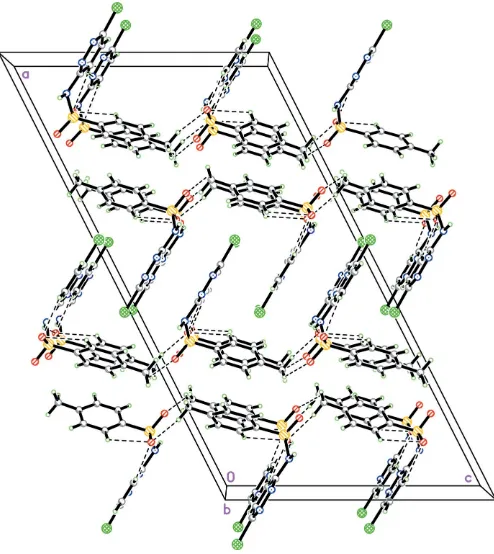
stabilized by intermolecular N—H O hydrogen bonds. Weak intramolecular C—H O and intermolecular C— H O and C—H N interactions are also observed. The molecules are linked into one-dimensional chains along thec
axis and these chains are interconnected, forming a two-dimensional network.
Comment
N-(6-Chloro-3-pyridazinyl)-4-methylbenzenesulfonamide, (I), is a synthetic antibacterial drug of the sulfanilamide family (Ciba Ltd, 1961). A number of sufanilamide drugs have been crystallographically characterized in recent decades (Acharya
et al., 1982; Adsmond & Grant, 2001; Basaket al., 1983; Caira & Mohamed, 1992; Deoet al., 1980; Joshiet al., 1983). Some of them, viz. sulfamerazine and sulfamethazine, have been studied several times (Acharyaet al., 1982; Adsmond & Grant, 2001; Basak et al., 1983; Cairaet al., 1992; Deo et al., 1980). Recently, we reported the crystal structure of sulfachloro-pyridazine (Tanet al., 2005) and we report here the structure of the title compound, (I), a chloropyridazine sulfonamide derivative.
The heterocyclic ring geometry in (I) is comparable to that found for free pyridazine (Blake & Rankin, 1991). The pyri-dazine and benzene rings form a distorted V configuration indicated by the torsion angle C5—S1—N1—C1 of 54.74 (18); the dihedral angle between these two rings is
73.79 (11), which is smaller than in sulfachloropyridazine [82.86 (6); Tanet al., 2005].
Weak intramolecular C2—H2A O1 and C10— H10A O1 interactions (Table 2) generate R1
2(6) and R 1 2(5)
motifs, respectively (Bernstein, et al., 1995). The inter-molecular hydrogen bond N1—H1N1 O1(x, 1 + y, z), involves the sulfonamide NH group and sulfonamide O atom. Molecules are linked into one-dimensional chains along thec
axis through weak C—H O interactions (Fig. 2 and Table 2). These chains are linked together through further weak C— H O interactions (Table 2), forming a two-dimensional network. A C—H interaction is also observed (Table 2,Cg
is the centroid of the benzene ring).
Experimental
N-(6-Chloropyridazin-3-yl)-4-methylbenzenesulfonamide (0.2 mmol) and Zn(CH3COO)2(0.5 mmol) were placed in a Pyrex tube. After addition of EtOH (1.0 ml) and H2O (0.5 ml), the tube was frozen with liquid N2, evacuated and sealed with a torch. The tube was heated at 343 K for 1 d to give light-yellow rod-shaped crystals of (I) in a 46% yield.
Crystal data
C11H10ClN3O2S
Mr= 283.74
Monoclinic,C2=c a= 29.8577 (4) A˚ b= 5.6198 (1) A˚ c= 16.1140 (2) A˚
= 116.892 (1) V= 2411.45 (7) A˚3
Z= 8
Dx= 1.563 Mg m
3
MoKradiation
= 0.49 mm1 T= 297 (2) K Rod, light yellow 0.500.330.27 mm
Data collection
Bruker SMART APEX2 CCD area-detector diffractometer
!scans
Absorption correction: multi-scan (SADABS; Bruker, 2005) Tmin= 0.792,Tmax= 0.881
19119 measured reflections 3507 independent reflections 2965 reflections withI> 2(I) Rint= 0.027
max= 30.0
Refinement
Refinement onF2
R[F2> 2(F2)] = 0.050
wR(F2) = 0.159
S= 1.05 3507 reflections 168 parameters
H atoms treated by a mixture of independent and constrained refinement
w= 1/[2(F
o2) + (0.0966P)2
+ 1.8315P]
whereP= (Fo2+ 2Fc2)/3
(/)max< 0.001
max= 0.73 e A˚3
min=0.50 e A˚
[image:2.610.43.295.72.212.2]3
Table 1
Selected geometric parameters (A˚ ,).
S1—O1 1.4258 (17) S1—O2 1.4345 (15) S1—N1 1.6592 (18) S1—C5 1.7373 (19) Cl1—C4 1.731 (2)
N1—C1 1.406 (2) N2—C1 1.317 (3) N2—N3 1.341 (3) N3—C4 1.304 (3)
O1—S1—O2 118.62 (10) O1—S1—N1 108.07 (10) O2—S1—N1 104.04 (10) O1—S1—C5 109.04 (10)
O2—S1—C5 109.91 (10) N1—S1—C5 106.41 (9) C1—N1—S1 122.74 (13)
O1—S1—N1—C1 62.25 (19) O2—S1—N1—C1 170.81 (17) C5—S1—N1—C1 54.74 (18) S1—N1—C1—C2 42.5 (3)
N1—S1—C5—C10 105.64 (16) O1—S1—C5—C6 170.73 (14) O2—S1—C5—C6 39.13 (18) N1—S1—C5—C6 72.93 (16)
organic papers
Acta Cryst.(2006). E62, o2178–o2180 Funet al. C
11H10ClN3O2S
o2179
Figure 1
[image:2.610.46.293.267.544.2]The molecular structure of (I), showing 50% probability displacement ellipsoids and the atomic numbering. Intramolecular hydrogen bonds are shown as dashed lines.
Figure 2
[image:2.610.314.566.593.717.2]Table 2
Hydrogen-bond geometry (A˚ ,).
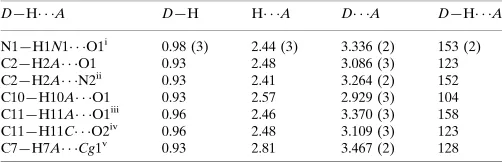
D—H A D—H H A D A D—H A
N1—H1N1 O1i
0.98 (3) 2.44 (3) 3.336 (2) 153 (2) C2—H2A O1 0.93 2.48 3.086 (3) 123 C2—H2A N2ii
0.93 2.41 3.264 (2) 152 C10—H10A O1 0.93 2.57 2.929 (3) 104 C11—H11A O1iii 0.96 2.46 3.370 (3) 158 C11—H11C O2iv
0.96 2.48 3.109 (3) 123 C7—H7A Cg1v
0.93 2.81 3.467 (2) 128
Symmetry codes: (i)x;yþ1;z; (ii)x;y1;z; (iii)x;y;z1
2; (iv)x;yþ1;z 1 2; (v)
xþ1 2;yþ
1 2;zþ
1
2.Cgis the centroid of the benzene ring.
The H atom bound to atom N1 was located in a difference Fourier map and refined isotropically. The remainding H atoms were placed in calculated positions, with C—H distances in the range 0.93–0.98 A˚ . TheUisovalues were constrained to be 1.5Ueqof the carrier atom for methyl H atoms and 1.2Ueqfor the remaining H atoms.
Data collection:APEX2(Bruker, 2005); cell refinement:APEX2; data reduction: SAINT (Bruker, 2005); program(s) used to solve structure: SHELXTL (Sheldrick, 1998); program(s) used to refine structure:SHELXTL; molecular graphics:SHELXTL; software used to prepare material for publication:SHELXTLandPLATON(Spek, 2003).
The National Natural Science Foundation of China (contract/grant Nos. 20361002 and 30460153) is thanked for its support. The authors also thank the Malaysian Government
and Universiti Sains Malaysia for the Scientific Advancement Grant Allocation (SAGA) grant No. 304/PFIZIK/653003/ A118.
References
Acharya, K. R., Kuchela, K. & Kartha, G. (1982).J. Cryst. Spectro. Res.12, 369–376.
Adsmond, D. A. & Grant, D. J. W. (2001).J. Pharm. Sci.90, 2058–2077. Allen, F. H., Kennard, O., Watson, D. G., Brammer, L., Orpen, A. G. & Taylor,
R. (1987).J. Chem. Soc. Perkin Trans. 2, pp. S1–19.
Basak, A. K., Mazumdar, S. K. & Chaudhuri, S. (1983).Acta Cryst.C39, 492– 494.
Bernstein, J., Davis, R. E., Shimoni, L. & Chamg, N.-L. (1995).Angew. Chem. Int. Ed. Engl.34, 1555–1573.
Blake, A. J. & Rankin, D. W. H. (1991).Acta Cryst.C47, 1933–1936. Bruker (2005).APEX2(Version 1.27),SAINT(Version 7.12a) andSADABS
(Version 2004/1). Bruker AXS Inc., Madison, Wisconsin, USA. Caira, M. R. & Mohamed, R. (1992).Acta Cryst.B48, 492–498. Ciba Ltd (1961). Br. Patent No. 884827.
Deo, N., Tiwari, R. K. & Singh, T. P. (1980).J. Sci. Res. (Bhopal, India),2, 137– 139.
Garcia-Raso, A., Fiol, J. J., Martorell, G., Lopez-Zafra, A. & Quiros, M. (1997). Polyhedron,16, 613–621.
Joshi, V. V., Tiwari, R. K., Patel, T. C. & Singh, T. P. (1983).India J. Phys. A,57, 79–89.
Sheldrick, G. M. (1998).SHELXTL. Version 5.1. Bruker AXS Inc., Madison, Wisconsin, USA.
Spek, A. L. (2003).J. Appl. Cryst.36, 7–13.
Tan, Y.-S., Chen, Z.-F., Liang, H. & Zhang, Y. (2005).Acta Cryst.E61, o1842– o1844.
Yuan, R.-X., Xiong, R.-G., Chen, Z.-F., Zhang, P., Ju, H.-X., Dai, Z., Guo, Z.-J., Fun, H.-K. & You, X.-Z. (2001).J. Chem. Soc. Dalton Trans.pp. 774–776.
organic papers
o2180
Funet al. Csupporting information
sup-1 Acta Cryst. (2006). E62, o2178–o2180
supporting information
Acta Cryst. (2006). E62, o2178–o2180 [https://doi.org/10.1107/S1600536806015832]
N
-(6-Chloropyridazin-3-yl)-4-methylbenzenesulfonamide
Hoong-Kun Fun, Shu-Feng Zhang, Zhen-Feng Chen, Hong Liang and Suchada Chantrapromma
N-(6-chloropyridazin-3-yl)-4-methylbenzenesulfonamide
Crystal data
C11H10ClN3O2S
Mr = 283.74
Monoclinic, C2/c
Hall symbol: -C 2yc
a = 29.8577 (4) Å
b = 5.6198 (1) Å
c = 16.1140 (2) Å
β = 116.892 (1)°
V = 2411.45 (7) Å3
Z = 8
F(000) = 1168
Dx = 1.563 Mg m−3
Mo Kα radiation, λ = 0.71073 Å Cell parameters from 2965 reflections
θ = 1.5–30.0°
µ = 0.49 mm−1
T = 297 K
Rod, colorless or light yellow? 0.50 × 0.33 × 0.27 mm
Data collection
Bruker SMART APEX2 CCD area-detector diffractometer
Radiation source: fine-focus sealed tube Graphite monochromator
Detector resolution: 8.33 pixels mm-1
ω scans
Absorption correction: multi-scan (SADABS; Bruker, 2005)
Tmin = 0.792, Tmax = 0.881
19119 measured reflections 3507 independent reflections 2965 reflections with I > 2σ(I)
Rint = 0.027
θmax = 30.0°, θmin = 1.5°
h = −42→41
k = −7→7
l = −22→22
Refinement
Refinement on F2
Least-squares matrix: full
R[F2 > 2σ(F2)] = 0.050
wR(F2) = 0.159
S = 1.05 3507 reflections 168 parameters 0 restraints
Primary atom site location: structure-invariant direct methods
Secondary atom site location: difference Fourier map
Hydrogen site location: inferred from neighbouring sites
H atoms treated by a mixture of independent and constrained refinement
w = 1/[σ2(F
o2) + (0.0966P)2 + 1.8315P]
where P = (Fo2 + 2Fc2)/3
(Δ/σ)max < 0.001
Δρmax = 0.73 e Å−3
supporting information
sup-2 Acta Cryst. (2006). E62, o2178–o2180
Special details
Experimental. The data was collected with the Oxford Cyrosystem Cobra low-temperature attachment.
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes.
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2,
conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used
only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2
are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger.
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
x y z Uiso*/Ueq
S1 0.149188 (17) 0.16291 (9) 0.34156 (3) 0.04022 (16) Cl1 −0.08085 (2) 0.45853 (12) −0.02156 (5) 0.0659 (2) O1 0.13103 (6) −0.0735 (3) 0.33793 (11) 0.0522 (4) O2 0.18807 (6) 0.2488 (3) 0.42799 (10) 0.0580 (4) N1 0.10183 (6) 0.3493 (3) 0.31781 (12) 0.0444 (4) N2 0.04107 (8) 0.5648 (3) 0.20208 (16) 0.0591 (5) N3 −0.00186 (8) 0.5919 (3) 0.12367 (17) 0.0638 (6) C1 0.05824 (7) 0.3496 (3) 0.23199 (13) 0.0376 (4) C2 0.03368 (8) 0.1437 (3) 0.18517 (15) 0.0457 (4) H2A 0.0461 −0.0068 0.2080 0.055* C3 −0.00945 (8) 0.1727 (4) 0.10440 (16) 0.0478 (5) H3A −0.0272 0.0431 0.0689 0.057* C4 −0.02557 (7) 0.4030 (4) 0.07782 (15) 0.0443 (4) C5 0.16706 (6) 0.1989 (3) 0.25361 (12) 0.0347 (3) C6 0.19286 (7) 0.4054 (3) 0.25216 (14) 0.0397 (4) H6A 0.2003 0.5210 0.2978 0.048* C7 0.20707 (7) 0.4362 (3) 0.18292 (15) 0.0416 (4) H7A 0.2243 0.5734 0.1821 0.050* C8 0.19603 (7) 0.2638 (3) 0.11340 (13) 0.0403 (4) C9 0.16950 (8) 0.0594 (4) 0.11517 (14) 0.0448 (4) H9A 0.1616 −0.0556 0.0692 0.054* C10 0.15498 (7) 0.0274 (3) 0.18414 (14) 0.0399 (4) H10A 0.1372 −0.1080 0.1845 0.048* C11 0.21237 (10) 0.2905 (4) 0.04729 (16) 0.0519 (5) H11A 0.1847 0.2684 −0.0133 0.078* H11B 0.2379 0.1745 0.0571 0.078* H11C 0.2260 0.4473 0.0516 0.078* H1N1 0.1152 (12) 0.504 (6) 0.346 (2) 0.070 (9)*
Atomic displacement parameters (Å2)
U11 U22 U33 U12 U13 U23
supporting information
sup-3 Acta Cryst. (2006). E62, o2178–o2180
O2 0.0456 (8) 0.0790 (12) 0.0333 (7) −0.0017 (8) 0.0036 (6) −0.0045 (7) N1 0.0372 (8) 0.0498 (10) 0.0432 (8) 0.0009 (7) 0.0155 (7) −0.0108 (7) N2 0.0507 (10) 0.0320 (8) 0.0713 (13) −0.0013 (7) 0.0071 (9) −0.0063 (8) N3 0.0501 (10) 0.0324 (9) 0.0822 (15) 0.0024 (7) 0.0064 (10) 0.0037 (9) C1 0.0333 (8) 0.0346 (8) 0.0438 (9) −0.0003 (6) 0.0166 (7) −0.0041 (7) C2 0.0417 (10) 0.0287 (8) 0.0536 (11) 0.0015 (7) 0.0100 (8) 0.0009 (7) C3 0.0430 (10) 0.0330 (9) 0.0535 (11) −0.0022 (7) 0.0095 (9) −0.0014 (7) C4 0.0354 (8) 0.0388 (9) 0.0516 (11) 0.0015 (7) 0.0134 (8) 0.0058 (8) C5 0.0297 (7) 0.0331 (8) 0.0354 (8) 0.0018 (6) 0.0096 (6) −0.0009 (6) C6 0.0362 (8) 0.0325 (8) 0.0456 (9) −0.0029 (6) 0.0144 (7) −0.0084 (7) C7 0.0376 (9) 0.0315 (8) 0.0525 (10) −0.0020 (6) 0.0175 (8) 0.0001 (7) C8 0.0387 (9) 0.0400 (9) 0.0400 (9) 0.0052 (7) 0.0159 (7) 0.0038 (7) C9 0.0519 (11) 0.0382 (9) 0.0410 (9) −0.0028 (8) 0.0181 (8) −0.0080 (7) C10 0.0407 (9) 0.0311 (8) 0.0427 (9) −0.0030 (6) 0.0144 (7) −0.0037 (6) C11 0.0617 (13) 0.0549 (12) 0.0474 (11) −0.0028 (10) 0.0321 (10) 0.0011 (9)
Geometric parameters (Å, º)
S1—O1 1.4258 (17) C5—C10 1.395 (2) S1—O2 1.4345 (15) C5—C6 1.399 (2) S1—N1 1.6592 (18) C6—C7 1.372 (3) S1—C5 1.7373 (19) C6—H6A 0.9300 Cl1—C4 1.731 (2) C7—C8 1.403 (3) N1—C1 1.406 (2) C7—H7A 0.9300 N1—H1N1 0.98 (3) C8—C11 1.366 (3) N2—C1 1.317 (3) C8—C9 1.403 (3) N2—N3 1.341 (3) C9—C10 1.375 (3) N3—C4 1.304 (3) C9—H9A 0.9300 C1—C2 1.395 (3) C10—H10A 0.9300 C2—C3 1.365 (3) C11—H11A 0.9600 C2—H2A 0.9300 C11—H11B 0.9600 C3—C4 1.379 (3) C11—H11C 0.9600 C3—H3A 0.9300
supporting information
sup-4 Acta Cryst. (2006). E62, o2178–o2180
C3—C2—C1 117.06 (18) C9—C10—C5 119.77 (18) C3—C2—H2A 121.5 C9—C10—H10A 120.1 C1—C2—H2A 121.5 C5—C10—H10A 120.1 C2—C3—C4 117.03 (18) C8—C11—H11A 109.5 C2—C3—H3A 121.5 C8—C11—H11B 109.5 C4—C3—H3A 121.5 H11A—C11—H11B 109.5 N3—C4—C3 124.3 (2) C8—C11—H11C 109.5 N3—C4—Cl1 115.05 (16) H11A—C11—H11C 109.5 C3—C4—Cl1 120.62 (16) H11B—C11—H11C 109.5 C10—C5—C6 120.26 (17)
O1—S1—N1—C1 −62.25 (19) O2—S1—C5—C10 142.30 (16) O2—S1—N1—C1 170.81 (17) N1—S1—C5—C10 −105.64 (16) C5—S1—N1—C1 54.74 (18) O1—S1—C5—C6 −170.73 (14) C1—N2—N3—C4 −0.8 (4) O2—S1—C5—C6 −39.13 (18) N3—N2—C1—C2 0.3 (4) N1—S1—C5—C6 72.93 (16) N3—N2—C1—N1 −176.3 (2) C10—C5—C6—C7 −1.3 (3) S1—N1—C1—N2 −141.00 (19) S1—C5—C6—C7 −179.83 (14) S1—N1—C1—C2 42.5 (3) C5—C6—C7—C8 0.1 (3) N2—C1—C2—C3 1.1 (3) C6—C7—C8—C11 −176.9 (2) N1—C1—C2—C3 177.3 (2) C6—C7—C8—C9 0.8 (3) C1—C2—C3—C4 −1.8 (3) C11—C8—C9—C10 177.1 (2) N2—N3—C4—C3 −0.1 (4) C7—C8—C9—C10 −0.6 (3) N2—N3—C4—Cl1 179.3 (2) C8—C9—C10—C5 −0.5 (3) C2—C3—C4—N3 1.4 (4) C6—C5—C10—C9 1.4 (3) C2—C3—C4—Cl1 −177.98 (18) S1—C5—C10—C9 180.00 (15) O1—S1—C5—C10 10.70 (18)
Hydrogen-bond geometry (Å, º)
D—H···A D—H H···A D···A D—H···A
N1—H1N1···O1i 0.98 (3) 2.44 (3) 3.336 (2) 153 (2)
C2—H2A···O1 0.93 2.48 3.086 (3) 123 C2—H2A···N2ii 0.93 2.41 3.264 (2) 152
C10—H10A···O1 0.93 2.57 2.929 (3) 104 C11—H11A···O1iii 0.96 2.46 3.370 (3) 158
C11—H11C···O2iv 0.96 2.48 3.109 (3) 123
C7—H7A···Cg1v 0.93 2.81 3.467 (2) 128