Copyright1999 by the Genetics Society of America
Analysis of Mutations in the Yeast mRNA Decapping Enzyme
Sundaresan Tharun and Roy Parker
Departments of Molecular and Cellular Biology and Biochemistry and the Howard Hughes Medical Institute, University of Arizona, Tucson, Arizona 85721-0106
Manuscript received September 29, 1998 Accepted for publication December 14, 1998
ABSTRACT
A major mechanism of mRNA decay in yeast is initiated by deadenylation, followed by mRNA decapping, which exposes the transcript to 59to 39exonucleolytic degradation. The decapping enzyme that removes the 59cap structure is encoded by the DCP1 gene. To understand the function of the decapping enzyme, we used alanine scanning mutagenesis to create 31 mutant versions of the enzyme, and we examined the effects of the mutations both in vivo and in vitro. Two types of mutations that affected mRNA decapping in vivo were identified, including a temperature-sensitive allele. First, two mutants produced decapping enzymes that were defective for decapping in vitro, suggesting that these mutated residues are required for enzymatic activity. In contrast, several mutants that moderately affected mRNA decapping in vivo yielded decapping enzymes that had at least the same specific activity as the wild-type enzyme in vitro. Combination of alleles within this group yielded decapping enzymes that showed a strong loss of function in vivo, but that still produced fully active enzymes in vitro. This suggested that interactions of the decapping enzyme with other factors may be required for efficient decapping in vivo, and that these particular mutations may be disrupting such interactions. Interestingly, partial loss of decapping activity in vivo led to a defect in normal dependent decapping, but it did not affect the rapid deadenylation-independent decapping triggered by early nonsense codons. This observation suggested that these two types of mRNA decapping differ in their requirements for the decapping enzyme.
T
HE turnover of mRNA is an important control to 39 exonuclease Xrn1p that degrades mRNA after point in the regulation of gene expression. Though decapping have been identified in several organisms mRNA decay mechanisms vary with organisms and the (Heyeret al. 1995;Bashkirov et al. 1997).nature of the mRNA, several commonalities do exist Decapping is a key step in the turnover of yeast among the various eukaryotes (Ross 1995; Jacobson mRNAs, because variation in decapping rates accounts andPeltz1996;TharunandParker1997). For exam- for part of the differences in decay rates of specific ple, in many eukaryotes, shortening of the poly(A) tail mRNAs in yeast (Muhlrad et al. 1994, 1995).
Decap-forms an initial step that is followed by the degradation ping also occurs in a process termed mRNA surveil-of the body surveil-of the mRNA (BeelmanandParker1995). lance, whereby mRNAs containing a premature trans-In Saccharomyces cerevisiae, mRNA deadenylation leads to lational stop codon are rapidly decapped and degraded decapping of the mRNA, which is followed by rapid 59to by a 59to 39exonucleolytic process. In this case, how-39exonucleolytic degradation of the mRNA (Muhlrad ever, decapping occurs independently of deadenyla-andParker1992;DeckerandParker1993;Hsuand tion (Muhlrad and Parker 1994). Thus, given the Stevens1993;Muhlradet al. 1994, 1995). Several ob- central role of decapping in mRNA decay, resolution servations suggest that decapping and 59 to 39 decay of the mechanisms by which mRNA decapping occurs may also occur in other eukaryotes. For example, full- and is controlled will be critical for understanding length mRNAs that are devoid of both the cap and most mRNA turnover.
of the poly(A) tail have been detected from murine Earlier studies have identified the yeast DCP1 gene as liver cells (Couttetet al. 1997). Similarly, mRNA decay encoding a decapping enzyme that is required for intermediates that are shortened at their 59ends have mRNA decay in vivo and sufficient for decapping activity been identified in both plant and animal cells (Limand in vitro (Beelmanet al. 1996;LaGrandeurandParker Maquat 1992; Higgs and Colbert 1994; Gera and 1998). This decapping enzyme cleaves capped mRNAs Baker 1998). Furthermore, homologs of the yeast 59 within the cap 59–59 triphosphate linkage to release m7GDP and a 59-monophosphate mRNA (Beelman et
al. 1996). In strains deleted for the DCP1 gene, many
mRNAs are stabilized as a result of a block in mRNA Corresponding author: Roy Parker, Departments of Molecular and
Cellular Biology and Biochemistry and the Howard Hughes Medical decapping (Beelmanet al. 1996). These mRNAs include Institute, Life Sciences South Building, East Lowell St., P.O. Box 210
stable and unstable mRNAs that undergo
deadenyla-106, University of Arizona, Tucson, AZ 85721-0106.
E-mail: [email protected] tion-dependent decapping, as well as mRNAs with early
Mutants dcp1-41 to -44 were made by combining mutations nonsense codons that are degraded by
deadenylation-of allele 17 or 4 with mutations deadenylation-of 19 or dcp1-independent decapping. These results indicate that the
25. The mutations of dcp1-17 and dcp1-4 occur close to the N decapping enzyme Dcp1p is required for most, if not terminus of Dcp1p, while the dcp1-19 and dcp1-25 mutations all, mRNA decapping in vivo. Elucidation of the func- affect sites closer to the C terminus of the Dcp1p sequence. Therefore, the combinations were made by exchanging the tion and regulation of Dcp1p is, therefore, important
region coding for the N-terminal portion of Dcp1p in vectors for understanding decapping.
pRP886 and pRP889 with those derived from pRP885 and In the present work, we used alanine-scanning
muta-pRP874 by restriction digestion and ligation.
genesis to create 31 different mutant versions of the RNA preparation and analysis:For steady-state RNA analysis, decapping enzyme, and we examined the effects of the cells were grown to midlog phase in SC–Trp minimal medium containing sucrose and galactose as carbon sources. For mutations both in vivo and in vitro. Two types of
muta-mRNA half-life determination by transcriptional repression, tions that affected mRNA decapping were identified.
cells were grown as described above to midlog and then shifted One set of mutations resulted in strong loss of function
to SC–Trp minimal medium with glucose as a carbon source. both in vivo and in vitro, indicating that they affected RNA preparation and analysis were done as described pre-specific amino acid residues important for enzymatic viously (Caponigroet al. 1993). Levels of full-length species and poly(G)→39 end fragments of MFA2pG and PGK1pG function. The second set of mutations resulted in
mod-mRNAs were determined by probing Northern blots with erate, or in some allelic combinations, strong loss of
59-32P end-labeled oligonucleotides [capable of hybridizing to function in vivo, but they failed to cause any loss of
both full-length species and poly(G)→39end fragments] spe-activity of the protein in vitro. This suggests that the cific for the respective mRNAs (CaponigroandParker1995) amino acid residues that were changed in these mutants and by quantitating the signal with a PhosphorImager (Molec-ular Dynamics). To determine the levels of CYH2 mRNA and may be required for some functionally important in vivo
CYH2 pre-mRNA, a random-primed cDNA probe capable of interactions of Dcp1p with other proteins that were not
specifically hybridizing to both of them was used, and quantita-present in the purified in vitro system. Our studies also
tion was done using a PhosphorImager, as described above. revealed that mutants with partial loss of function in MFA2pG mRNA half-life measurements were performed by
vivo were not defective for the rapid deadenylation- the method of transcriptional repression from GAL1 UAS by
independent decapping triggered by early nonsense co- glucose, as described before (Parker et al. 1991). For each time point, the time of freezing (in dry ice) the cell pellet dons, and that they were defective only for the normal
was noted and used for drawing the decay curve. deadenylation-dependent decapping in vivo, suggesting
Construction of the FLAG-Dcp1p overexpression vector: a difference between these two processes with regard The FLAG-fused chimeras of mutant DCP1 genes were created to their requirement for the decapping enzyme. by PCR amplification of the mutant DCP1 sequences from their corresponding CEN vector clones (pRP872–pRP898, see above and Table 1) using the oligonucleotide GCAGCACCG GATC CATGGACTACAAGGAC GACGATGACAAGATGAC C MATERIALS AND METHODS
GGAGCAGCAAC (oRP311), which places the FLAG-coding sequence 59of the DCP1 gene, as well as the oligonucleotide Oligonucleotide-directed mutagenesis:All mutants of DCP1,
except dcp1-41 to -44, were generated by this procedure. For GCAGCACCGTCGACTTCTCACTTGGGCATCTC (oRP312). This PCR fragment was digested with BamHI and Sal I, and it performing site-directed mutagenesis, the DCP1 gene was
cloned into the yeast shuttle vector pUN45 (Elledge and was ligated into the yeast expression vector pG-1 (Schenaet al. 1991) between the BamHI and SalI sites. This way, FLAG fusion
Davis 1988), which contains the M13 origin of replication.
pUN45 is a CEN vector with the yeast TRP1 gene as an auxotro- chimeras of the alleles dcp1-2 (pRP900), dcp1-4 (pRP901), 7 (pRP902), 17 (pRP903), 19 (pRP904), dcp1-phic marker. The entire DCP1 gene present in the plasmid
p424DCP1 (Beelman et al. 1996) was excised as an ≈1.7-kb 25 (pRP905), and dcp1-31 (pRP906) were cloned. These 2m plasmids (with the TRP1 marker) express FLAG-Dcp1p from fragment by digesting p424DCP1 with enzymes ApaI and NotI,
and it was then inserted into the pUN45 vector digested with the constitutive glyceraldehyde-3-phosphate dehydrogenase promoter.
the same two enzymes. This resulted in the plasmid pRP783,
which was used for making all the mutants. The single- Purification of FLAG-Dcp1p:Wild-type and mutant FLAG-Dcp1p proteins were purified from dcp1D cells (yRP1071) stranded form of this plasmid was made after transforming it
into Escherichia coli XL1 Blue cells and infecting them with carrying the plasmid pRP801 (LaGrandeur and Parker
1998) and the 2m clones of the mutant DCP1 genes (see M13K07 helper phage. Site-directed mutagenesis was then
performed following standard methods by annealing muta- above), respectively. A 200-ml culture of the yRP1071 cells harboring the appropriate 2mplasmid was grown to late-log genic oligonucleotide to the single-stranded form of pRP783,
extending it with Klenow to complete the second strand, and phase in SC–Trp minimal medium containing 2% glucose, and it was harvested by centrifugation. Cells were washed in closing the second strand with ligase. After this, a portion of
the mutagenesis reaction mixture was transformed into E. coli buffer A (10 mmTris-HCl, pH 7.6, 100 mmpotassium acetate, 2 mmmagnesium acetate, and 2 mm2-mercaptoethanol), spun TB1 cells, and plasmid minipreps made from several
trans-formants were screened by restriction analysis (using pRP783 down, and suspended in 1.5 ml/g cell pellet weight of the same buffer containing COMPLETE (Boehringer Mannheim, as control) to find those bearing the mutated DCP1 gene.
This yielded plasmids pRP872–pRP898, which bore the various Indianapolis, IN) protease inhibitors. An equal volume of acid-washed glass beads was added to the suspension, and cells primary mutant forms of DCP1 (Table 1). These plasmids were
then transformed into dcp1Dstrain yRP1071 (Beelmanet al. were lysed by six cycles of vortexing for 15 and 45 sec of cooling in ice water. The lysate was centrifuged at 18,0003g 1996), and the transformants were studied for their RNA
TABLE 1
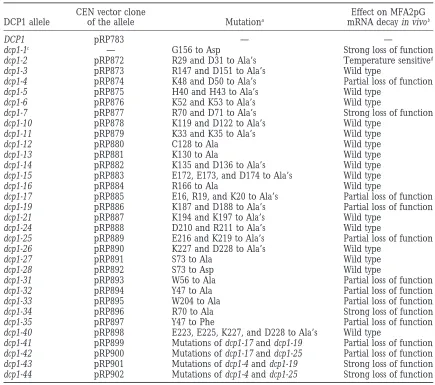
Amino acid residues changed in the variousdcp1alleles and the phenotype of the mutants
CEN vector clone Effect on MFA2pG
DCP1 allele of the allele Mutationa mRNA decay in vivob
DCP1 pRP783 — —
dcp1-1c — G156 to Asp Strong loss of function
dcp1-2 pRP872 R29 and D31 to Ala’s Temperature sensitived
dcp1-3 pRP873 R147 and D151 to Ala’s Wild type
dcp1-4 pRP874 K48 and D50 to Ala’s Partial loss of function
dcp1-5 pRP875 H40 and H43 to Ala’s Wild type
dcp1-6 pRP876 K52 and K53 to Ala’s Wild type
dcp1-7 pRP877 R70 and D71 to Ala’s Strong loss of function
dcp1-10 pRP878 K119 and D122 to Ala’s Wild type
dcp1-11 pRP879 K33 and K35 to Ala’s Wild type
dcp1-12 pRP880 C128 to Ala Wild type
dcp1-13 pRP881 K130 to Ala Wild type
dcp1-14 pRP882 K135 and D136 to Ala’s Wild type
dcp1-15 pRP883 E172, E173, and D174 to Ala’s Wild type
dcp1-16 pRP884 R166 to Ala Wild type
dcp1-17 pRP885 E16, R19, and K20 to Ala’s Partial loss of function
dcp1-19 pRP886 K187 and D188 to Ala’s Partial loss of function
dcp1-21 pRP887 K194 and K197 to Ala’s Wild type
dcp1-24 pRP888 D210 and R211 to Ala’s Wild type
dcp1-25 pRP889 E216 and K219 to Ala’s Partial loss of function
dcp1-26 pRP890 K227 and D228 to Ala’s Wild type
dcp1-27 pRP891 S73 to Ala Wild type
dcp1-28 pRP892 S73 to Asp Wild type
dcp1-31 pRP893 W56 to Ala Partial loss of function
dcp1-32 pRP894 Y47 to Ala Partial loss of function
dcp1-33 pRP895 W204 to Ala Partial loss of function
dcp1-34 pRP896 R70 to Ala Strong loss of function
dcp1-35 pRP897 Y47 to Phe Partial loss of function
dcp1-40 pRP898 E223, E225, K227, and D228 to Ala’s Wild type
dcp1-41 pRP899 Mutations of dcp1-17 and dcp1-19 Partial loss of function
dcp1-42 pRP900 Mutations of dcp1-17 and dcp1-25 Partial loss of function
dcp1-43 pRP901 Mutations of dcp1-4 and dcp1-19 Strong loss of function
dcp1-44 pRP902 Mutations of dcp1-4 and dcp1-25 Strong loss of function
aTotal of 27 primary mutations changing 43 residues. This includes 38 charged residues, two Trp’s, and one each of Ser, Cys, and Tyr.
bAs judged from the steady-state level of poly(G)→39 end fragment in relation to that of the full-length species.
cThe dcp1-1 allele was identified earlier (Hatfieldet al. 1996) by screening mutants generated by ethyl methanesulfonate mutagenesis.
dFor MFA2pG mRNA decay, dcp1-2 is wild type at 188, but almost completely inactive at 368.
antibody immuno-affinity column (Kodak) equilibrated with assay: Uncapped mRNAs lacking poly(A) tails were synthe-sized in vitro by T7 RNA polymerase runoff transcriptions. Full-buffer A. The flowthrough was collected and reapplied to the
column. The column was sequentially washed with 15 bed length MFA2 mRNA was transcribed from plasmid pRP802
(LaGrandeur and Parker 1998) that was linearized with
volumes each of buffer AN (buffer A with 0.05% NP-40), buffer
ANS (buffer AN with 0.7mpotassium acetate), and buffer AN. EcoRI to produce a 343-nucleotide mRNA consisting of 325 nucleotides of MFA2 sequence, followed by 18 nucleotides FLAG-Dcp1p was eluted with 5 bed volumes of buffer AN
con-taining 200mg/ml FLAG peptide. The FLAG peptide present encoded by the plasmid polylinker.
T7 transcriptions were done in 100-ml reactions containing in the FLAG-Dcp1p preparation was removed by dialysis, and
the FLAG-Dcp1p was finally stored in 20 mmTris, pH 7.5, 1–2mg of template DNA, 5 mmNTPs, 40 mmTris-HCl, pH 8.0, 1 mmspermidine, 5 mm DTT, 50 mg/ml BSA, 0.01% with 5 mmDTT and 20% (v/v) glycerol.
The FLAG-Dcp1p preparation was analyzed by standard Triton X-100, 20 mmMgCl2, 5 units yeast inorganic pyrophos-phatase (Sigma, St. Louis, MO) and 40 units T7 RNA polymer-SDS-PAGE methods on a 10% gel (Laemmli1970). Protein
size markers were purchased from GIBCO BRL (Gaithersburg, ase (Boehringer Mannheim) at 378forz15 hr (Harriset al. 1994). The uncapped transcripts were purified by polyacryl-MD). Silver staining of SDS-PAGE gels was done using the
Silver Stain Plus kit from Bio-Rad (Richmond, CA) following amide electrophoresis.
7-Methyl caps were added to in vitro-synthesized MFA2 tran-the manufacturer’s protocol.
[a-32P]GTP, 40 units RNasin (Promega, Madison, WI), 0.67 mmS-adenosylmethionine, 50 mmTris-HCl, pH 7.6, 2 mm
MgCl2, 6 mmKCl, 1 mmDTT, and 25 units guanylyltransferase (GIBCO-BRL) at 378for 2 hr (Beelmanet al. 1996). As a result of the qualitative nature of capping by guanylyltransferase, these conditions typically capped only 35–40% of the RNA in the reaction (Shuman and Moss 1990). Cap-labeled RNAs were separated from unincorporated label by Sepharose-CL6B (Pharmacia, Piscataway, NJ) spin chromatography.
Decapping assays:Decapping assays used in this study were similar to those described previously (Beelman et al. 1996). m7G[32P]pppMFA2 mRNA was incubated with Dcp1p prepara-tion in 50 mmHEPES, pH 7.0, containing 1 mmMgCl2, 0.05% NP-40, and 1 mmDTT in a total reaction volume of 15ml at 308. Reactions were stopped by addition of EDTA to a final 50-mmconcentration and shifting the sample to ice. To follow the reaction time course, aliquots of the reaction mixture were drawn at different time points, mixed with EDTA, and placed on ice. The product of the reaction (a-32P-m7GDP) was sepa-rated from the unreacted substrate by PEI-cellulose thin-layer chromatography developed in 0.45m(NH4)2SO4. Activity at
any time t is calculated by dividing the amount of product Figure1.—Positions (on the amino acid sequence) of the formed at time t by the total amount of substrate taken for various primary mutations of DCP1 generated in this study. the reaction (the sum of the amount of product formed and The amino acid sequence of Dcp1p is shown. Underlines the amount of unreacted substrate at time t). The activity indicate the parts of Dcp1p sequence that are targeted in values were then normalized for the amount of Dcp1p protein different primary mutants, and the actual residues mutated in the sample to determine the specific activity values, which are in boldface letters. The allele number of each mutant were used to plot graphs for the time courses. Quantitations of (preceded by #) is given below the corresponding underlines. product formed and unreacted substrate left after the reaction All amino acid changes are to alanines, except where indicated were done using a Molecular Dynamics PhosphorImager. To (two cases, dcp1-28 and dcp1-35). The numbers shown above determine the amount of Dcp1p protein in the purified prepa- the sequence are the amino acid residue numbers starting rations, Western blot of purified samples was probed with from the N terminus. The allele dcp1-1 (Gly156 to Asp, shown anti-Dcp1p antibodies, and the Dcp1p band intensity in the in italics) was isolated earlier (Hatfieldet al. 1996) by screen-autoradiograph was then quantitated using the IP Lab Gel ing mutants generated by ethyl methanesulfonate mutagenesis. program. In addition, for careful comparison, protein
concen-trations of the purified enzymes were adjusted and then the activity was reassayed where equal amounts of purified Dcp1p
Mutants dcp1-41 to -44 were made by different pairwise could be compared directly (see Figure 6 for example).
When-ever a set of purified Dcp1p samples were compared for spe- combinations of the mutations of dcp1-4, -17, -19, and cific activity, they were all assayed together with wild-type -25 by restriction digestion and ligation (seematerials Dcp1p using the same substrate preparation. Assays done with
and methodsand Table 1). In most (26 out of 31) of different substrate preparations were not compared.
the mutants, #3 residues were changed. Five residues each were affected in dcp1-41 and -42, and four each
RESULTS were changed in dcp1-40, -43, and -44 (Table 1). In each
case, it was confirmed by sequencing that the mutant Mutagenic strategy:For the analysis of Dcp1p, we used
DCP1 gene contained only the targeted mutations. The
site-directed mutagenesis to change residues that are
positions of all mutations on the primary sequence are expected to be important for function. Two approaches
shown in Figure 1. were used. First, we used the strategy of
charged-to-Effects of the mutations on deadenylation-dependent alanine scanning mutagenesis. In this procedure,
clus-decappingin vivo:To study the effect of the mutations ters of charged amino acids in the primary sequence,
in the DCP1 gene, we determined if they affected mRNA which are likely to be on the surface of the folded
pro-decapping in vivo. Loss of pro-decapping enzyme function tein, were identified and systematically changed to
ala-(as in dcp1D) leads to a block in mRNA decay in vivo nines (Wells 1991). In the second approach, we
tar-(Beelmanet al. 1996) because the 59→39exonucleolytic geted some residues that might be important on the
digestion of mRNA in vivo (by Xrn1p) is dependent on basis of comparison to other proteins that interact with
the removal of mRNA cap by the DCP1 gene product. the cap site. For example, because the cap structure
The dcp1 mutant alleles were expressed from a centro-is often recognized by a stacking interaction between
mere plasmid in the dcp1Dyeast strain yRP1071 ( Beel-aromatic residues (Hodelet al. 1997;Marcotrigiano
manet al. 1996). The ability of the different dcp1 mutants et al. 1997), we targeted the aromatic residues Trp56,
to degrade mRNAs was assessed by estimating the decay Trp204, and Tyr47 of DCP1. All mutants except
dcp1-of both the unstable MFA2pG and stable PGK1pG
41 to -44 were made by standard oligomutagenesis on
mRNAs in vivo. Both of these mRNAs are known to a copy of the DCP1 gene cloned in a centromere plasmid
leading to 59to 39digestion by the exonuclease Xrn1p (Muhlrad et al. 1994, 1995). In the yRP1071 strain,
the MFA2pG and PGK1pG genes (driven by the GAL promoter) are expressed from a chromosomal location, and they contain a poly(G) tract insertion in their 39 untranslated regions. The poly(G) tract does not affect the decay of these transcripts, and it serves to block 59 to 39 exonucleolytic degradation in cis (Decker and Parker1993). As a result, when the decapped mRNA is degraded by Xrn1p, it leads to the accumulation of a fragment of the mRNA which lacks the part of mRNA on the 59side of the poly(G) stretch as a degradation intermediate, termed the poly(G)→39end fragment. In the absence of any decapping, however, the action of Xrn1p and, hence, the formation of a mRNA fragment resulting from the poly(G) tract is blocked (Beelman et al. 1996). Thus, the relative levels of the full-length
species and poly(G)→39 end fragment of the mRNA can be used as a first approximation of the efficiency of decapping and 59to 39exonucleolytic digestion (see Hatfieldet al. 1996). Given this, we examined the
full-length to poly(G)→39 end fragment ratios for the MFA2pG and PGK1pG transcripts in all the dcp1 mutants. The results of these studies (summarized in Table 1) are as follows. First, 16 of the dcp1 primary mutations (covering 27 residues) did not have any significant effect on the ratio of poly(G)→39end fragment to full-length mRNA and, hence, were not pursued further. These
alleles include dcp1-3, -5, -6, -10, to -16, -21, -24, -26 to Figure 2.—Half-life of MFA2pG mRNA in different dcp1 -28 and -40. Second, the mutant dcp1-7 showed a large primary mutants. Shown in the figure are MFA2pG mRNA half-life measurements in different dcp1 mutants (grown at effect on the ratio. In this mutant, Arg70 and Asp71
308) on Northern blots after transcriptional shutoff by shifting were both converted to alanines (Figure 1). Subsequent
galactose-induced cells to glucose medium. The time points studies indicated that the severity of the phenotype of
at which cells were collected for RNA isolation are indicated this mutant could be almost fully attributed to the muta- on top of the gel pictures. The name of the dcp1 allele and tion of Arg70. This was revealed by the mutant dcp1- the MFA2pG half-life determined are shown on the right of each gel picture. Northern analysis using an oligonucleotide
34, wherein only Arg70 is mutated to alanine. Third, 9
probe specific for MFA2pG mRNA and half-life determination mutants, dcp1-2, -4, -17, -19, -25, -31, -32, -33, and -35,
was done as described inmaterials and methods.
showed a partial change in the poly(G)→39end frag-ment to full-length mRNA ratio, suggesting that these
alleles represented partial loss-of-function mutations. poly(G)→39 end fragment. As expected, mutants with Importantly, it should be noted that the dcp1 mutations a large change in the ratio (e.g., dcp1-34) showed a large affected both MFA2pG and PGK1pG mRNAs similarly change in t1/2, and the partial loss-of-function mutants in each case, indicating that mRNA-specific rates of de- showed a more modest effect. Thus, these observations capping cannot be attributed to the decapping enzyme indicate that we have identified several mutations in the
per se (seediscussion). decapping enzyme that affect mRNA turnover in vivo.
Figure 3.—Temperature-sen-sitive RNA decay phenotype of alleles dcp1-2 and dcp1-34. Wild-type, dcp1D, dcp1-2, and dcp1-34 cells were grown at the tempera-tures indicated above the lanes in galactose-containing minimal medium, and RNA made from them was analyzed by Northern analysis using an oligonucleo-tide probe specific for MFA2pG mRNA, as described in materi-als and methods.
34 allele also showed small differences in function in pressed at levels comparable to the wild-type enzyme. Western analysis of crude extracts made from the vari-response to the temperature of growth. This allele was
essentially like a null allele at 308 and higher, but it ous mutant dcp1 strains performed with Dcp1p anti-bodies showed that the level of the Dcp1p protein was produced a very small amount of the poly(G)→39end
fragment at 188 (Figure 3). These thermosensitive al- not substantially different in the mutants compared to wild-type strain, with the exception of the dcp1-25 mu-leles, especially dcp1-2, should be useful for the analysis
of mRNA decay (e.g., seeJacobs AndersonandParker tant, which showed reduced levels compared to wild type (data not shown). This observation indicated that 1998).
In vitro analysis of the mutant decapping enzymes: the loss-of-function phenotypes of the mutants, with the exception of dcp1-25, were not caused by changes in The above studies identified a number of mutations in
Dcp1p that led to a defect in decapping in vivo. In protein stability and were, therefore, likely to be caused principle, these defects could arise by several means, by the mutations’ effects on either the enzymatic func-including destabilizing the protein, altering the enzy- tion of the decapping enzyme or its ability to interact matic properties of the decapping enzyme, or pre- with other proteins required for decapping in vivo. venting interactions with other factors required for To determine if the mutant Dcp1ps expressed in these proper decapping. To distinguish these possibilities, we alleles were defective in enzymatic activity, we purified first determined if the mutant proteins were being ex- them and assayed their ability to decap a capped mRNA
in vitro. For this purpose, we expressed FLAG
epitope-tagged versions of wild-type and mutant dcp1 genes from
Figure4.—Half-life of MFA2pG mRNA at different temper-atures in the allele dcp1-2. Shown is the MFA2pG mRNA half-life measurement in wild-type and dcp1-2 cells grown at 188
and 368 and in dcp1Dcells (carrying only the pUN45 vector Figure 5.—SDS-PAGE analysis of various purified FLAG-Dcp1p samples: FLAG-tagged decapping enzyme was purified without insert) grown at 368on Northern blots after
transcrip-tional shutoff by shifting galactose-induced cells to glucose from wild-type cells and dcp1 mutant cells using anti-FLAG antibody affinity column, subjected to SDS-PAGE, and silver medium. The time points at which cells were collected for
RNA isolation are indicated on top of the gel pictures. Allele stained as described in materials and methods.The dcp1 allele, from which the protein was purified, is shown on top name, growth temperature, and the MFA2pG half-life
deter-mined are shown on the right of each gel picture. Northern of each lane. The Dcp1p band is indicated by an arrow on the left. The positions of molecular weight markers are shown analysis using an oligonucleotide probe specific for MFA2pG
mRNA and half-life determination were done as described in on right. The slight differences seen in the mobilities of some of the mutant proteins are not reproducible.
Figure6.—In vitro decapping activity of FLAG-tagged forms of wild-type and mutant Dcp1p proteins. Mutant and wild-type Dcp1p proteins were expressed as FLAG fusion proteins from the constitutive GPD promoter in a 2mvector (seematerials and methods) in a dcp1Dbackground and were purified using FLAG antibody column, as described inmaterials and methods.
Purified proteins were assayed in vitro for decapping activity, as described inmaterials and methods. (A) Time course of decapping reaction carried out with wild-type and mutant FLAG-tagged decapping enzymes. Aliquots of the reaction mixture were drawn at 7, 14, and 25 min, resolved by TLC after stopping the reaction, and activity was quantitated and normalized for protein amounts, as described inmaterials and methods.(B) Phosphorimage of a typical TLC run. Decapping reactions were carried out with the purified wild-type and mutant FLAG-Dcp1p samples for 20 min, after which the reaction was stopped and TLC was performed. The position of the product of the reaction, m7GDP and the origin where the unreacted substrate stays in the TLC plate are indicated by arrows on the right side of the phosphorimage. Shown below the TLC picture is a Western analysis of the FLAG-Dcp1p samples used in the assay (slight differences seen in the mobilities of some of the mutant proteins are not reproducible). The FLAG-Dcp1p samples used for the Western analysis and the assay are indicated below the lanes of the Western blot. For every FLAG enzyme sample, the amounts used for the assay shown in the TLC picture and the Western analysis are equal.
2mvectors (seematerials and methods) in dcp1Dcells zyme for the substrate. Figure 6A shows time course curves of decapping reactions performed with wild-type (strain yRP1071). FLAG-tagged proteins encoded by
the following alleles were purified (seematerials and and mutant FLAG-Dcp1p samples drawn by plotting decapping activity values that were normalized for the methods): wild type; five of the mutant alleles that lead
to partial defects in mRNA decapping in vivo (dcp1-17, amount of Dcp1p protein present in the respective samples, as described in materials and methods. dcp1-4, dcp1-19, dcp1-25, and dcp1-31); the allele dcp1-7,
which is completely defective in RNA decay in vivo; and Assays were also done after adjusting the protein con-centrations of the individual preparations so that equal the allele dcp1-2, which has a temperature-sensitive RNA
decay phenotype in vivo. Analysis of the purified protein amounts of dcp1 protein were compared directly (Figure 6B). The results are summarized in Table 2.
samples on SDS polyacrylamide gel revealed a clean
z30-kD protein (Figure 5) that was verified to be Dcp1p Two classes of mutant proteins were identified. First, the alleles dcp1-2 and dcp1-7, which caused a severe loss by Western analysis (Figure 6B). The purified enzymes
were then assayed in vitro for decapping activity using of mRNA decay function in vivo, yielded FLAG fusion proteins that had in vitro-specific activities at least five
in vitro-synthesized MFA2 mRNA labeled with32P in the
cap. Upon decapping, this substrate releases radiola- times less than that of wild-type FLAG-Dcp1p (Table 2 and Figure 6). We interpreted this observation to indi-beled m7 GDP, which is separated from it by TLC (see
materials and methods). These assays were done un- cate that the dcp1-2 and dcp1-7 mutations alter residues that are important for the enzymatic function of Dcp1p. der limiting concentrations of substrate so that they
TABLE 2
Relative specific activities of purified wild-type and mutant Dcp1p proteins
Specific activity
Protein (% wild type)a
Dcp1p 100
Dcp1-17p 104
Dcp1-4p 291
Dcp1-19p 101
Dcp1-25p 289
Dcp1-7p 18
Dcp1-2p 18
Dcp1-43p 165
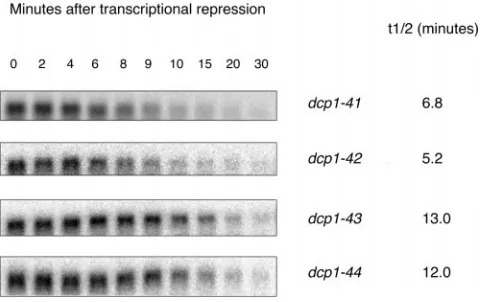
Figure7.—Half-life of MFA2pG mRNA in mutants dcp1-41
Dcp1-44p 241
to -44. Shown are MFA2pG mRNA half-life measurements in
Dcp1-31p 103
alleles dcp1-41 to -44 (grown at 308) on Northern blots after aRelative specific activities were calculated from the 7-min transcriptional shutoff by shifting galactose-induced cells to time point values of the experiment shown in Figure 6A. See glucose medium. The time points at which cells were collected
materials and methodsfor details of procedures. for RNA isolation are indicated on top of the gel pictures.
The name of the dcp1 allele and the MFA2pG half-life deter-mined are shown on the right of each gel picture. Northern analysis using an oligonucleotide probe specific for MFA2pG a partial loss of function in vivo, 4, 17,
dcp1-mRNA and half-life determination was done as described in
19, dcp1-25, and dcp1-31. Unlike their in vivo phenotypes,
materials and methods.
the in vitro-specific activities of the decapping enzymes made by these mutants did not show any loss of function compared to the wild-type protein. As seen in Table 2
combination of the lesions should yield proteins with and Figure 6, all the mutant proteins had specific
activi-strong defects in vivo, but still active in vitro when the ties equal to or greater than that of wild-type Dcp1p.
enzyme is purified. This result was confirmed in multiple preparations of
To this end, mutations of dcp1-17 and dcp1-4 alleles the proteins. The higher specific activity (compared to
were individually combined by restriction digestion and that of wild-type protein) of some of the mutant proteins
ligation with mutations of dcp1-19 or dcp1-25. This re-may result from the fact that when Dcp1p is
overex-sulted in four new mutants, dcp1-41 (combination of pressed, only a fraction of the enzyme is active (
LaGran-dcp1-17 and dcp1-19 mutations), dcp1-42 (combination deur and Parker 1998), and it is possible that these
of dcp1-17 and dcp1-25 mutations), dcp1-43 (combination mutants alter the percentage of enzyme that is active,
of dcp1-4 and dcp1-19 mutations), and dcp1-44 (combina-possibly as a result of the defect in the ability to decap
tion of dcp1-4 and dcp1-25 mutations). The plasmids mRNAs. Nevertheless, the clear and important result is
were then individually transformed into dcp1Dstrain to that these proteins are active decapping enzymes in
analyze their phenotypes. isolation, yet they show defects in decapping in vivo.
MFA2pG mRNA half-life was measured in the mutant One possible explanation for the mutations that affect
strains dcp1-41 to -44. As shown in Figure 7, the alleles decapping in vivo but not in vitro is that they target
dcp1-41 and -42 (combination of mutations of dcp1-17
residues required for interactions in vivo that are absent
with those of dcp1-19 and dcp1-25, respectively) do not in the in vitro experiment. Alternatively, because these
cause any significantly stronger loss of function than partial loss-of-function mutants have only a weak RNA
the primary partial loss-of-function mutants and, hence, decay phenotype (moderate stabilization of MFA2pG
were not studied further. On the other hand, the other mRNA) in vivo, another explanation could be that the
two alleles, dcp1-43 and dcp1-44 (combination of muta-effects of these mutations on the in vitro enzymatic
effi-tions of dcp1-4 with those of dcp1-19 and dcp1-25, respec-ciency of the protein is too small to be detected. One
tively) clearly resulted in a much higher stabilization of way to distinguish between these two possibilities would
MFA2pG mRNA than any of the primary partial loss-of-be to combine the mutations borne by two or more
function mutants. In addition, Western analysis showed of these partial loss-of-function mutants to make new
that all these mutant proteins were expressed at approxi-mutants that show a more severe loss of function in
mately the same levels as the wild-type protein (data not
vivo and to study the specific activity of the decapping
shown). enzyme made by such mutants. If the second possibility
To find if the mutant Dcp1p made in the dcp1-43 were true, then one would predict these proteins to
and dcp1-44 mutants was active in vitro, we made FLAG have significantly less specific activity than the wild-type
epitope-tagged versions of the two mutant dcp1 genes protein. On the other hand, if these mutants affect
veillance: The decapping enzyme Dcp1p functions in both the normal deadenylation-dependent decay path-way and in the deadenylation-independent decapping triggered by early nonsense codons (Beelman et al.
1996). The experiments described above examined the effects of dcp1 mutations on the former process (because MFA2pG and PGK1pG mRNAs decay by this pathway). Therefore, we wanted to examine the effects of the dcp1 mutations on the deadenylation-independent decap-ping process. For this purpose, we took advantage of the observation that the CYH2 pre-mRNA is a poorly spliced RNA; some unspliced precursor enters the cyto-plasm in wild-type cells (He et al. 1993). Because the
intronic sequence leads to an in-frame premature termi-nation codon, this pre-mRNA forms a natural substrate for the deadenylation-independent decapping pathway, which requires Dcp1p (He et al. 1993; Beelman et al.
1996). Thus, the accumulation of CYH2 premRNA in
vivo can be used as a measure of how efficiently the
deadenylation-independent decapping mechanism op-erates in the cells.
Examination of the extent of CYH2 pre-mRNA accu-mulation by Northern analysis in the various dcp1 mu-tants is shown in Figure 8. As expected, in wild-type cells, very little CYH2 pre-mRNA accumulated in relation to the CYH2 mRNA. In contrast, the amount of the CYH2 pre-mRNA accumulated in the dcp1Dstrain and in all the strong loss-of-function mutants, except for dcp1-43 (i.e., in dcp1-2, -7, -34, and -44), was substantially higher than in wild-type cells. Consistent with their MFA2pG mRNA decay phenotype in vivo, dcp1-7 and -34 showed higher CYH2 pre-mRNA accumulation than did dcp1-2 and -44. The allele dcp1-43 caused only a moderate in-crease in CYH2 premRNA accumulation compared to
Figure 8.—Effect of the different dcp1 mutations on the
wild-type cells. Nevertheless, the dcp1 alleles that had degradation of CYH2 pre-mRNA in vivo. RNA made from
wild-type and mutant cells grown to midlog at 308 in minimal a partial loss of function in deadenylation-dependent medium was subjected to Northern analysis using a random- decay (i.e., MFA2pG mRNA decay) did not show any primed CYH2 cDNA probe (which hybridizes to both CYH2
significant increase in the accumulation of CYH2 pre-pre-mRNA and mRNA), as described inmaterials and
meth-mRNA compared to the wild-type control. It is
impor-ods.(A) Phosphorimages of Northern blots. The dcp1 allele
tant to note here that as seen in Figure 8, the levels of from which RNA was prepared is indicated above each lane,
and positions of the CYH2 pre-mRNA and mRNA are shown CYH2 pre-mRNA are very low in wild-type cells, and they on the left side of gel picture. (B) Ratio of the level of CYH2 are increased about fourfold in dcp1Dcells, suggesting pre-mRNA to that of CYH2 mRNA in different dcp1 alleles
that z75% of the pre-mRNA pool in wild-type cells determined by quantitation of the corresponding bands in
decays in a DCP1-dependent manner. the phosphorimages shown in A.
The observation that the partial loss-of-function muta-tions did not affect deadenylation-independent decap-ping could be explained in two ways. First, these muta-them as described earlier. In vitro decapping assays
per-formed with these mutant proteins revealed that both tions could affect some specific structural feature(s) of Dcp1p that is required only for deadenylation-depen-of these mutant proteins had specific activities similar
to those of the wild-type protein (Table 2 and Figure dent decapping. This possibility seemed unlikely be-cause similar results were seen with all the partial loss-6). This strongly supports the idea that the amino acid
residues changed in these mutants are not likely to be of-function alleles. Alternatively, the rate of decapping of the nonsense-containing mRNAs (which undergo important for the enzymatic activity of Dcp1p, but,
rather, are important for some functionally important deadenylation-independent decapping) could be less sensitive (than normal mRNAs that undergo deadenyla-interactions of Dcp1p with other proteins in vivo.
sur-Figure 9.—Decay of non-sense mRNA substrate in vivo is not sensitive to modest losses of Dcp1p function. (A) Effect of growth temperature on the steady-state ratio of the levels of full-length (FL) species and poly(G)→39 end fragment of MFA2pG mRNA and of pre-mRNA and pre-mRNA of CYH2 in a dcp1-2 mutant in vivo. RNA was made from wild-type cells (filled circles) grown at 188, 308, and 368 and from dcp1-2 cells (open circles) grown at 188, 208, 228, 248, 278, 308, 338, and 368 and was analyzed by Northern using an end-labeled oligonucleotide probe specific for MFA2pG. Blots were then stripped and reprobed with a random-primed CYH2 cDNA probe. Steady-state levels of full-length species and poly(G)→ 39 end fragments of MFA2pG mRNA and pre-mRNA and mRNA of CYH2 were deter-mined as described in materi-als and methods, and ratios were calculated. (B) Accumulation of CYH2 pre-mRNA at steady state at 188, 308, and 368in vivo in a dcp1-34 strain, which shows severe loss of function at 188and complete loss of function at 308and 368for MFA2pG mRNA decay in vivo. Wild-type and dcp1-34 cells were grown at the temperatures indicated above the lanes to midlog in minimal medium, and RNA made from them was analyzed by Northern analysis using CYH2 cDNA probe (which hybridizes to both CYH2 pre-mRNA and mRNA) as described
inmaterials and methods.
possibility, we took advantage of the temperature sensi- and, therefore, a partial decrease in decapping activity affects only normal mRNA decay function. This idea tivity of the dcp1-2 allele. The logic was to determine if
there would be a differential effect on nonsense-medi- was further supported by studying the CYH2 pre-mRNA accumulation in the other mutant with temperature-ated vs. regular mRNA decay as we raised the
tempera-ture and, thereby, decreased the proportion of func- sensitive MFA2pG mRNA decay phenotype, dcp1-34. In this mutant, the accumulation of CYH2 pre-mRNA is tional decapping enzyme in vivo.
For this purpose, we prepared RNA from the dcp1-2 almost as low as in the wild-type control at 188(Figure 9B), while there is a strong defect in the decapping of mutant strain grown at 188, 208, 228, 248, 278, 308, 338,
and 368, and from wild-type cells grown at 188, 308, and the MFA2pG transcript in vivo at the same temperature (as shown by very low levels of poly(G) fragment in 368, and we determined the effects on normal
decap-ping (by examination of the full-length to fragment relation to full-length MFA2pG mRNA in Figure 3). Importantly, Figure 9 shows that at 368, the difference ratios for the MFA2pG transcript) and
deadenylation-independent decapping (by examination of the CYH2 in CYH2 pre-mRNA/mRNA ratio between wild-type and
dcp1-2 cells is approximately fourfold, which is
compara-pre-mRNA accumulation relative to CYH2 mRNA
accu-mulation). Comparison of these data showed that decay ble to the approximately fourfold difference observed in that ratio between wild-type and dcp1D cells grown of the CYH2 pre-mRNA was less sensitive to decreases
in the levels of decapping activity. The critical observa- at 308(see Figure 8). Because dcp1-2 cells are almost as defective in decapping as dcp1Dcells at 368(see Figure tion was that in dcp1-2 mutants, the decay of MFA2pG
mRNA was significantly defective at temperatures where 4), this indicates that the size of the CYH2 pre-mRNA pool degrading in a DCP1-dependent fashion is not dras-the CYH2 pre-mRNA decay remains normal (Figure
9A). For example, at 308, there was a substantial increase tically altered at higher temperatures. We have also ob-served a similar fold difference in this ratio when wild-in the amount of full-length MFA2pG transcript relative
to the poly(G)→39 end fragment, whereas there was type cells and dcp1Dcells grown at 368were compared (data not shown).
little change in the amount of CYH2 pre-mRNA
accumu-lation. This observation was consistent with the explana- As mentioned earlier (Figure 8), the strong loss-of-function allele, dcp1-43, caused only a slight increase in tion that in vivo, the decapping activity is limiting for
wild type, unlike all the other strong loss-of-function methyltransferase have been shown to interact with the cap structure by stacking the aromatic ring of 7-methyl alleles, dcp1-7, -34, -2, and -44, which resulted in a
sub-stantial increase in accumulation of the CYH2 pre- guanine between the ring structures of their conserved aromatic amino acid residues, and that the methyl mRNA. At the present time, we do not know if this is
because the mutations borne by this allele (dcp1-43) group specificity has at least partly been attributed to such a stacking interaction (Hodelet al. 1997; Marco-affect features of the decapping enzyme that are
specifi-cally required for deadenylation-dependent decapping trigiano et al. 1997;Matsuo et al. 1997). This raises
the possibility that these aromatic residues in yeast (normal mRNA decay). Further work will be needed to
resolve this issue. Dcp1p may have similar functions.
In the case of Tyr47, its importance for the protein’s function may arise either by virtue of its aromatic ring DISCUSSION
or its hydroxyl, as in RNase-A, where a conserved Tyr residue has been shown to be required for stabilizing Identification of residues important for Dcp1p
activ-ity:In this work, we have identified several residues that the active site structure (Eberhardtet al. 1996) by
hydro-gen bond interactions through its hydroxyl. Therefore, are important for the ability of the decapping enzyme
to function. The strongest effects were seen with the we tested the effect of removing only the hydroxyl mo-iety of the Tyr47 in the mutant dcp1-35, where Tyr47 is mutation of arginine at position 70 (dcp1-34) and with
the dcp1-2 mutant, in which Arg29 and Asp31 were both replaced with Phe. The fact that this mutation, like
dcp1-32, also causes a partial loss of function is consistent
changed to alanine residues. The dcp1-34 mutant was
defective in decapping in vivo at all temperatures tested, with the idea of the hydroxyl of Tyr47 being important (Figure 2). This suggests that the Tyr47 hydroxyl is likely with only a very small amount of mRNA turnover
ob-served at 188. The dcp1-2 mutant was more strikingly to be engaged in an important hydrogen bond interac-tion.
temperature sensitive in its function and showed little
difference from the wild type at a low temperature, yet An interesting observation was that the Dcp1p puri-fied from the partial loss-of-function mutants (dcp1-17, it was phenotypically similar to a null mutant at 368. In
addition to their strong in vivo phenotypes, both dcp1-2 dcp1-4, dcp1-19, dcp1-25, and dcp1-31) had
wild-type-spe-cific activity in vitro. This was also true for the mutants and dcp1-7 produced proteins that were defective in
enzymatic activity when purified. This observation indi- dcp1-43 and dcp1-44, which contained combinations of
alleles that led to a strong defect in decapping in vivo, cated that these residues were critical for the enzyme’s
function. Interestingly, both Arg70 and Asp31 are con- yet produced a functional decapping enzyme in vitro. These observations indicated that these lesions alter a served among a family of related ORFs in the database
with homology to the DCP1 sequence (S. Mian and property of the decapping enzyme that is not assayable in the current in vitro system. One formal possibility R. Parker, unpublished data). One potential role for
these conserved arginine and aspartic residues is to be is that these mutations alter the enzyme’s structure/ folding in such a manner that its catalytic efficiency is involved in recognition of the substrate. This possibility
is suggested by studies on the cytoplasmic cap-binding lost in vivo but not in vitro. This possibility seems un-likely, considering that this effect was seen with seven protein and the viral cap recognition protein, which
show that Arg and Asp residues are involved in recogniz- different mutants. An alternate and simpler explanation is that these lesions disrupt activation (or recruitment ing the cap moiety. For example, in both proteins, acidic
amino acid residues form hydrogen bonds with the N1 onto the substrate) of Dcp1p by some other factors that are required for efficient decapping in vivo rather than and N2 positions of the 7-methylguanine through their
side chain carboxyl oxygens (Hodelet al. 1997;Marco- by affecting Dcp1p’s own catalytic effeciency. Such acti-vation may involve the direct interaction of such fac-trigianoet al. 1997). Similarly, thea- andb-phosphate
oxygens of m7GDP interact with an Arg and a Lys resi- tors with Dcp1p. The importance of the requirement of other gene products for the functioning of Dcp1p due of eIF4E (Marcotrigiano et al. 1997). Thus, in
Dcp1p, Arg29, Asp31, and Arg70 could have analogous in vivo has also been suggested by earlier work in which
mutations in other genes that affect decapping in vivo roles. Alternatively, these two residues could play an
important but not absolutely required role in the cata- without altering the levels of the decapping enzyme were identified (Hatfieldet al. 1996;Boecket al. 1998).
lytic mechanism of the decapping enzyme.
A second set of interesting residues are the aromatic Moreover, the specific activity of Dcp1p was found to be significantly higher when it is a part of the crude residues, Trp56, Trp204, and Tyr47, each of which
causes a partial loss of function when changed to ala- lysate rather than in purified form (T. E. LaGrandeur andR. Parker,unpublished observations).
nine. These residues are also conserved among the
fam-ily of related ORFs in the database with homology to Interestingly, among the four mutants that were made by combining mutations borne by different pairs of
pri-DCP1. Furthermore, the importance of such aromatic
primary partial loss-of-function mutants) loss-of-func- 34 cells showed a strong defect in MFA2pG mRNA decay
tion phenotype in vivo, while the other two (dcp1-41 and at 188 (Figure 3), but they showed no effect on CYH2
dcp1-42) were defective roughly to the same extent as premRNA accumulation at this temperature (Figure 9B). the primary partial loss-of-function mutants. In other The above observations argue that nonsense-medi-words, only two combinations (out of four) of the pri- ated decapping is less sensitive to perturbations in the mary mutations resulted in aggravating the loss-of-func- decapping enzyme’s function than normal deadenyla-tion phenotype of the primary mutadeadenyla-tions while the other tion-dependent decapping. This suggests that the non-two did not. This could happen if the interaction of sense codon-containing substrate may be a more easily Dcp1p with other factor(s) involves several sites of con- accessible substrate for decapping than the normal tact on the Dcp1p molecule and some of these contacts mRNA substrate because of the manner in which decap-are redundant with each other. In that case, in any given ping is triggered in response to a nonsense codon. This primary partial loss-of-function mutant—where a given view has two implications. First, other trans-acting muta-site(s) is already mutated—introducing additional mu- tions that have been described as being specific for tations in sites that are redundant (for interaction) with deadenylation-dependent decapping (Hatfield et al.
that will fail to aggravate the phenotype any further (for 1996;Boecket al. 1998) may in fact simply lead to partial
discussion of a similar situation see Holtzman et al. loss-of-function phenotypes for decapping activity, with 1994). Further work will be required to determine the the ensuing effect only on normal mRNAs. Thus, the proteins that interact with Dcp1p and if those interac- MRT1, MRT3, and SPB8 gene products may not truly tions are indeed affected by these lesions. be specific for deadenylation-dependent decapping per The effects of Dcp1p mutations on differential se. The second interesting implication is that by limiting mRNA decapping rates:An important question is how decapping, either in cis or in trans, a situation could the different rates of mRNA decapping are specified on be created where a nonsense-mediated decay will still individual mRNAs. In principle, there could be specific occur, but deadenylated mRNAs will be stable. Strik-interactions with the decapping enzyme that recruit the ingly, this situation describes the state in Xenopus oo-enzyme to individual mRNAs at different rates. From cytes wherein normal mRNAs are stable as deadenylated this perspective, it would be expected that specific alleles species, yet nonsense-containing mRNAs are still rap-of the Dcp1p would affect mRNAs differentially. In this idly degraded (Whitfieldet al. 1994). This implies that light, we initially compared the effects of the mutations modulation of the levels of the decapping activity, either on the decay of the PGK1, a stable mRNA, and MFA2, in cis or in trans, may be important in early development an unstable mRNA. Both of these mRNAs degrade by and in other biological situations.
the deadenylation-dependent decapping pathway, and the difference in their decay rates at least partly results from the difference in their decapping rates (Decker
LITERATURE CITED
et al. 1993; Muhlrad et al. 1994, 1995). The fact that
Bashkirov, V. I., H. Scherthan, J. A. Solinger, J.-M. Buerstedde none of our mutations show a differential effect on the
andW.-D. Heyer,1997 A mouse cytoplasmic exoribonuclease
decay of these mRNAs suggests that DCP1 per se is
un-(mXRN1p) with preference for G4 tetraplex substrates. J. Cell
likely to distinguish between these two transcripts. This Biol. 136: 761–773.
observation is also consistent with more recent work Beelman, C. A.,and R. Parker,1995 Degradation of mRNA in eukaryotes. Cell 81: 179–183.
arguing that the status of the translation initiation
com-Beelman, C. A., A. Stevens, G. Caponigro, T. E. Lagrandeur, L. plex per se plays an important role in modulating decap- Hatfieldet al., 1996 An essential component of the decapping ping rates (LaGrandeur and Parker 1999; D. enzyme required for normal rates of mRNA turnover. Nature
382:642–646.
Schwartz and R. Parker, unpublished results; D.
Boeck, R., B. Lapeyre, C. E. BrownandA. B. Sachs,1998 Capped
MuhlradandR. Parker,unpublished results). mRNA degradation intermediates accumulate in the yeast spb8-2 Our experiments show that moderate losses of the mutant. Mol. Cell. Biol. 18: 5062–5072.
Caponigro, G., andR. Parker, 1995 Multiple functions for the
decapping enzyme’s function specifically affected only
poly(A)-binding protein in mRNA decapping and deadenylation
deadenylation-dependent decapping and did not have in yeast. Genes Dev. 9: 2421–2432.
any detectable effect on nonsense-mediated decay in Caponigro, G., D. MuhlradandR. Parker,1993 A small segment of MATa1 transcript promotes mRNA decay in Saccharomyces
cere-vivo. This conclusion was supported by the following
visiae : a stimulatory role for rare codons. Mol. Cell. Biol. 13: observations, in which conditions that led to a partial 5141–5148
defect in normal (MFA2pG) mRNA decay resulted in Couttet, P., M. Fromont-Racine, D. Steel, R. Pictet and T. Grange,1997 Messenger RNA deadenylation precedes
decap-no defect in decap-nonsense-mediated mRNA decay. First, in
ping in mammalian cells. Proc. Natl. Acad. Sci. USA 94: 5628–5633.
all the partial loss-of-function mutants studied, there
Decker, C. J.,andR. Parker,1993 A turnover pathway for both
was no increase in the accumulation of CYH2 pre-mRNA stable and unstable mRNAs in yeast: evidence for a requirement for deadenylation. Genes Dev. 7: 1632–1643.
compared to wild type (Figure 8). Second, with
increas-Eberhardt, E. S., P. K. Wittmayer, B. M. TemplerandR. T. Raines, ing growth temperature of dcp1-2 cells, MFA2pG mRNA
1996 Contribution of a tyrosine side chain to ribonuclease A
decay becomes defective at a lower temperature than catalysis and stability. Prot. Sci. 5: 1697–1703.
Elledge, S. J.,andR. W. Davis,1988 A family of versatile
dcp1-meric vectors designed for use in the sectoring-shuffle mutagene- Lim, S. K.,andL. E. Maquat,1992 Humanb-globin mRNAs that sis assay in Saccharomyces cerevisiae. Gene 70: 303–312. harbor a nonsense codon are degraded in murine erythroid
Gera, J. F.,andE. J. Baker,1998 Deadenylation-dependent and -inde- tissues to intermediates lacking regions of exon I or exons I and pendent decay pathways for alpha1-tubulin mRNA in Chlamydomo- II that have a cap-like structure at the 59termini. EMBO J. 11:
nas reinhardtii. Mol. Cell. Biol. 18: 1498–1505. 3271–3278.
Harris, M. E., J. M. Nolan, A. Malhotra, J. W. Brown, S. C. Harvey Marcotrigiano, J., A.-C. Gingras, N. SonenbergandS. K. Burley, et al., 1994 Use of photoaffinity crosslinking and molecular mod- 1997 Cocrystal structure of the messenger RNA 59cap-binding eling to analyze the global architecture of ribonuclease P RNA. protein (eIF-4E) bound to 7-methyl GDP. Cell 89: 951–961. EMBO J. 13: 3953–3963. Matsuo, H., H. Li, A. M. McGuire, C. M. Fletcher, A. C. Gingras
Hatfield, L., C. A. Beelman, A. StevensandR. Parker,1996 Muta- et al., 1997 Structure of translation factor eIF4E bound to tions in trans-acting factors affecting mRNA decapping in Saccharo- m7GDP and interaction with 4E-binding protein. Nat. Struct.
myces cerevisiae. Mol. Cell. Biol. 16: 5830–5838. Biol. 4: 717–724.
He, F., S. W. Peltz, J. L. Donahue, M. RosbashandA. Jacobson, Muhlrad, D., and R. Parker, 1992 Mutations affecting stability
1993 Stabilization and ribosome association of unspliced pre- and deadenylation of the yeast MFA2 transcript. Genes Dev. 6: mRNAs in a upf1Dmutant. Proc. Natl. Acad. Sci. USA 90: 7034– 2100–2111.
7038. Muhlrad, D.,andR. Parker,1994 Premature translational
termina-Heyer, W. D., A. W. Johnson, U. ReinhartandR. D. Kolodner, tion triggers mRNA decapping. Nature 370: 578–581.
1995 Regulation and intracellular localization of Saccharo- Muhlrad, D., C. J. DeckerandR. Parker,1994 Deadenylation of myces cerevisiae strand exchange protein 1 (Sep1/Xrn1/Kem1), a the unstable mRNA encoded by the yeast MFA2 gene leads to multifunctional exonuclease. Mol. Biol. Cell. 15: 2728–2736. decapping followed by 59to 39digestion of the transcript. Genes
Higgs, D. C.,andJ. T. Colbert,1994 Oat phytochrome A mRNA Dev. 8: 855–866. degradation appears to occur via two distinct pathways. Plant Cell
Muhlrad, D., C. J. DeckerandR. Parker,1995 Turnover
mecha-6:1007–1019. nisms of the stable yeast PGK1 mRNA. Mol. Cell. Biol. 15: 2145–
Hodel, A., X. ShiandP. D. Gershon,1997 Specific protein
recog-2156. nition of an mRNA cap through its alkylated base. Nat. Struct.
Parker, R., S. W. Peltz, D. HerrickandA. Jacobson,1991 Mea-Biol. 4: 350–354.
surement of mRNA decay rates in S. cerevisiae. Methods Enzymol.
Holtzman, D. A., K. F. WertmanandD. G. Drubin,1994 Mapping
194:415–422. actin surfaces required for functional interactions in vivo. J. Cell
Ross, J.,1995 mRNA stability in mammalian cells. Microbiol. Rev. Biol. 126: 423–432.
59:423–450.
Hsu, C. L.,andA. Stevens,1993 Yeast cells lacking 59→39
exoribo-Schena, M., D. PicardandK. R. Yamamoto,1991 Vectors for consti-nuclease 1 contain mRNA species that are poly (A) deficient and
tutive and inducible gene expression in yeast. Methods Enzymol. partially lack the 59cap structure. Mol. Cell. Biol. 13: 4826–4835.
194:389–398.
Jacobs Anderson, J. S.,andR. Parker,1998 The 39to 59
degrada-Shuman, S., andB. Moss, 1990 Purification and use of vaccinia tion of yeast mRNAs is a general mechanism for mRNA turnover
virus messenger RNA capping enzyme. Methods Enzymol. 181: that requires the SKI2 DEVH box protein and 39 to 59
exo-170–180. nucleases of the exosome complex. EMBO J. 17: 1497–1506.
Tharun, S.,andR. Parker,1997 Mechanisms of mRNA turnover
Jacobson, A.,andS. W. Peltz,1996 Interrelationships of the
path-in eukaryotic cells, pp. 181–199 path-in mRNA Metabolism and Post-ways of mRNA decay and translation in eukaryotic cells. Annu.
transcriptional Gene Regulation (Modern Cell Biology, Vol. 17),
Rev. Biochem. 65: 693–739.
edited byJ. B. HarfordandD. R. Morris. John Wiley Publishers,
Laemmli, U. K.,1970 Cleavage of structural proteins during the
assembly of the head of bacteriophage T4. Nature 227: 680–685. New York.
LaGrandeur, T. E.,andR. Parker,1998 Isolation and characteriza- Wells, J. A.,1991 Systematic mutational analyses of protein-protein tion of Dcp1p, the yeast mRNA decapping enzyme. EMBO J. 17: interfaces. Methods Enzymol. 202: 391–411.
101–110. Whitfield, T. T., C. R. SharpeandC. C. Wylie,1994
Nonsense-LaGrandeur, T. E.,andR. Parker,1999 The cis-acting sequences mediated mRNA decay in Xenopus oocytes and embryos. Dev. responsible for the differential decay of the unstable MFA2 and Biol. 165: 731–734.
stable PGK1 transcripts in yeast include the context of