Abstract
Chasse, Tyson Lee. Structural Effects on Encapsulation as Probed in Solution - Based and
Surface - Confined Redox-Active Core Dendrimers. (Advised by Professor Christopher B.
Gorman)
The purpose of this research was to study structure – property relationships of iron sulfur
core [Fe4S4(S-Dend)4]2- dendrimers. Previous studies have demonstrated that biasing dendrimer architecture increases the effective encapsulation of redox-active, paramagnetic,
Fe4S4 clusters. To further examine structure-property relationships of iron-sulfur core
dendrimers, studies were carried out to 1) probe the relationship between dendritic
architecture and encapsulation via the study of solution-based and surface-confined
constitutional isomers differing only in their benzyl substitution patterns, and 2) studying the
effects of counterion concentration and permeability on the electronic properties of
iron-sulfur core dendrimer thin films.
Three pairs of isomeric, iron-sulfur core dendrimers were synthesized. Each isomer pair was
distinguished by a 3,5-aromatic substitution pattern (extended) versus 2,6-aromatic
substitution pattern (backfolded). Several observations were made supporting the hypothesis
that the iron-sulfur cluster cores were encapsulated more effectively in the backfolded
microenvironment. Furthermore, heterogeneous electron-transfer rates for the backfolded
molecules were attenuated compared to the extended molecules. From diffusion
measurements obtained by pulsed field gradient spin-echo NMR and chronoamperometry,
the backfolded dendrimers were found to be smaller than the extended dendrimers.
Comparison of longitudinal proton relaxation (T1) values also indicated a smaller, more
compact dendrimer conformation for the backfolded architectures. These findings indicated
that dendrimer size was not the major factor in determining electron-transfer rate. Instead,
the effective electron-transfer distance, determined by the relative core position and mobility,
is most relevant for encapsulation.
In addition to solution studies, the electrochemical behavior of thin films composed of
redox-active, iron-sulfur core dendrimers were studied as a function of the type of counterion
available during reduction and re-oxidation. The rate of permeation/migration of counterions
into the film appeared to be the bottleneck to electron transfer through the film. As the
dendrimer is essentially non-polar, decreasing the relative polarity of the counterion
Biography
Acknowledgements
First and foremost, I would like to acknowledge my family and friends. It was the
friendships, love and guidance that you gave me throughout my life that has helped me get
where I am today. You have all touched my life in some way and I am a better person for
having had all of you in my life. Unfortunately, I can’t include everyone, but there are a few
who deserve a special thank you.
My friends that I met here at NC State were directly proportional to my sanity throughout
graduate school. They were always there to talk, whether it was science related or not, and
made life more enjoyable. The Saturday morning football crew was my first social
interaction and turned out to be where I met a lot of my good friends. Scot Bodnar and I
were true “Hall Ball” competitors and helped relieve the stress of chemistry and research. I
achieved a new level of witty banter that I never thought possible from daily conversations
with Ryan Fuierer. Finally, a thanks to my lab mate in Chris Cameron for our debates about
sports and science.
I truly have been blessed to find a best friend in Marcie Tinkham, who I was lucky enough to
marry. Her support and patience has been more than I ever thought possible. My education
and future has not come without sacrifices along the way, but Marcie never looked back on
indebted to her. I love you and I thank you for always being there for me as a wife and as a
friend.
My grandparents will always have a special place in my heart. They were a large part of my
childhood and taught me life lessons that I will always live by. Their love and support has
been incredible even though my future has taken me 1,000 miles away from them. It was
difficult moving away, but I always knew that they were happy and proud of me no matter
where I ended up.
Last but definitely not least, my parents. My father passed away when I was young but not
before he instilled in me the importance of being a great person. His attention to detail taught
me to put forth my best effort into everything I do. This was a great lesson not only in my
science career, but in my daily life. I was fortunate enough to have a stepfather in William
Allen. He was not only a father figure to me but also a great friend. He has really defined
the meaning of perseverance by earning a B.S. degree while working a full time job and still
being a wonderful family man. And finally, I would like to thank my mom. I have the
utmost respect for her and her ability to be a single parent and continuously steer me in the
right direction. She always wanted the best for and from me. Her love and support has
shaped me into the man I am today. Without her guidance, I can be sure that you would not
I acknowledge Dr. Chris Gorman for many lessons learned about being a professional as well
as a scientist. His guidance throughout my graduate career has been extremely helpful.
From multiple chats about research and science, to discussions about Maine and “lobstah’s,”
he has been not only an advisor but also a colleague. He has had a great influence on my
Table of Contents
LIST OF FIGURES
LIST OF TABLES
LIST OF SCHEMES
1. DENDRITIC ENCAPSULATION: AN ELECTRONIC
PROPERTIES PERSPECTIVE
1.1 ORIGIN OF DENDRIMERS
1.2 DENDRIMER SYNTHESIS: CONVERGENT VS. DIVERGENT 1.3 DENDRIMER ENCAPSULATION
1.3.1. DETECTION OF ENCAPSULATION: PHOTOPHYSICAL PROPERTIES
1.3.2. DETECTION OF ENCAPSULATION: ELECTROCHEMICAL PROPERTIES
1.4 DENDRIMER APPLICATIONS 1.4.1. ELECTRONIC MATERIALS 1.4.2. BIOLOGICAL MODELS
1.5 IRON-SULFUR (FE4S4) CORE DENDRIMERS
1.6 PERSPECTIVES OF DISSERTATION RESEARCH 1.7 REFERENCES
2. SYNTHESIS AND CHARACTERIZATION OF
CONSTITUTIONAL ISOMERS OF BENZYL-ETHER
IRON-SULFUR CORE DENDRIMERS
2.1 INTRODUCTION
2.2 RESULTS AND DISCUSSION
2.2.1. GENERAL SYNTHETIC METHODS 2.2.1.1. General Method for Halogenation
General Method for Chlorination (SOCl2)
General Method for Chlorination (CCl4/PPh3)
General Method for Bromination (CBr4/PPh3)
General Method for Bromination (PBr3)
2.2.1.2. General Method for Coupling Reaction
2.2.1.4. General Method for Deprotection 2.2.1.5. General Method for Ligand Exchange 2.3 EXPERIMENTAL COMPOUND DATA
2.4 REFERENCES
3. ELUCIDATION OF PHYSICAL PROPERTIES OF
CONSTITUTIONAL ISOMER IRON-SULFUR CORE
DENDRIMERS IN SOLUTION
3.1 INTRODUCTION
3.2 RESULTS AND DISCUSSION
3.2.1. DETERMINATION OF THERMODYNAMIC REDOX POTENTIAL AND KINETICS OF HETEROGENEOUS ELECTRON-TRANSFER
3.2.1.1. Cyclic Voltammetry
3.2.1.2. Osteryoung Square Wave Voltammetry
3.2.2. MOLECULAR SIZE AND ITS RELATION TO ELECTRON-TRANSFER RATE
3.2.2.1. Chronoamperometry
3.2.2.2. Pulsed Field Gradient Spin-Echo Nuclear Magnetic Resonance 3.2.3. FURTHER PROBING OF SIZE AND MOBILITY VIA NMR
RELAXATION MEASUREMENTS 3.2 CONCLUSION
3.3 EXPERIMENTAL
3.4.1. DETAILS OF THE ELECTROCHEMICAL ANALYSIS 3.4.1.1. Cyclic Voltammetry
3.4.1.2. Osteryoung Square Wave Voltammetry 3.4.1.3. Chronoamperometry
3.4.1.4. Pulsed Field Gradient Spin-Echo 1H-NMR
3.4.1.5. Inversion Recovery (T1 Relaxation Measurements) 3.4 REFERENCES
4. ELUCIDATION OF REDOX-PROPERTIES OF
CONSTITUTIONAL ISOMER IRON-SULFUR CORE
DENDRIMER THIN FILMS
4.1 INTRODUCTION
4.2 RESULTS AND DISCUSSION 4.2.1. THIN FILM PREPARATION
4.2.2. THIN FILM VS. SOLUTION COMPARISON
4.2.3. BACKFOLDED VS. EXTENDED THIN FILM COMPARISON 4.2.4. DETERMINATION OF KINETIC PROPERTIES
4.3 EXPERIMENTAL
4.3.1. MATERIALS/CHEMICALS
4.3.2. PREPARATION OF FILMS AND ELECTRODES 4.3.3. ELECTROCHEMICAL ANALYSIS
4.3.3.1. Apparatus
4.3.3.2. Electroanalysis of Films 4.4 REFERENCES
5. THE EFFECTS OF COUNTERION ON THE RATE OF
ELECTRON TRANSFER IN DENDRIMER THIN FILMS
5.1 INTRODUCTION
5.2 RESULTS AND DISCUSSION
5.2.1. SYSTEMATIC COUNTERION STUDY 5.2.1.1. Excluding Electrolyte Pre-Soak 5.2.1.2. Including Electrolyte Pre-Soak
5.2.2. ELECTROLYTE IONIC STRENGTH EFFECTS 5.3 CONCLUSION
5.4 EXPERIMENTAL
5.4.1. MATERIALS/CHEMICALS
5.4.2. PREPARATION OF FILMS AND ELECTRODES 5.4.3. ELECTROCHEMICAL ANALYSIS
5.4.3.1. Apparatus 5.4.3.2. Film Thickness
5.4.3.3. Electroanalysis of Films 5.5 REFERENCES
List of Figures
CHAPTER 1
FIGURE 1.1 ILLUSTRATION OF DENDRIMER PRIMARY STRUCTURE. COMPONENTS ARE SHOWN WITH PROPER RELATIVE
ARRANGEMENT AND ORIENTATION
FIGURE 1.2 DIVERGENT (LEFT) AND CONVERGENT (RIGHT)
DENDRIMER SYNTHETIC SCHEMATICS. GRAPHIC FROM FRECHET ET AL
FIGURE 1.3 ILLUSTRATION OF ELECTRON TRANSFER BETWEEN REDOX ACTIVE MOLECULES (LEFT) AND REDOX ACTIVE MOLECULES SURROUNDED (ENCAPSULATED) BY ORGANIC MATERIAL (RIGHT)
FIGURE 1.4 ILLUSTRATION OF THE IDEALOGICAL USE OF DENDRIMERS AS CHARGE TRAPPING UNITS TO CREATE A UNIMOLECULAR
BINARY MEMORY SYSTEM. UNENCAPSULATED REDOX
MOLECULES (LEFT) ALLOW CHARGE TRANSFER BETWEEN SITES, SCRAMBLING INFORMATION, WHILE ENCAPSULATING DENDIMERS TRAP CHARGE CREATING A BINARY SYSTEM OF 0’S (UNCHARGED) AND 1’S (CHARGED)
FIGURE 1.5 GENERATIONS 0-4 OF “FLEXIBLE”
4,4’-BIS(HYDROXYPHENYL)PENTANOL REPEAT UNIT DENDRIMERS (G0 – G4 FLEX)
FIGURE 1.6 GENERATIONS 0-4 OF “RIGID” 3,5-DISUBSTITUTED
GID) PHENYLACETYLENE REPEAT UNIT DENDRIMERS (G0 – G4 RI
FIGURE 1.7 HETEROGENEOUS ELECTRON TRANSFER RATES FOR THE “FLEXIBLE” AND “RIGID” DENDRIMERS PLOTTED AS A FUNCTION OF MOLECULAR WEIGHT
FIGURE 1.8 HETEROGENEOUS ELECTRON TRANSFER RATES FOR THE “FLEXIBLE” AND “RIGID” DENDRIMERS PLOTTED AS A FUNCTION OF THE MOLECULAR RADIUS OF GYRATION
CHAPTER 2
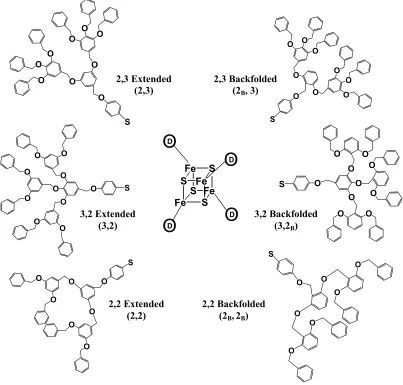
FIGURE 2.1 STRUCTURES OF THE CONSTITUTIONAL ISOMERIC DENDRIMER PAIRS. EACH DENDRIMER HAS THE FORM
(nBu4N)2{Fe4S4D4}, WHERE D INDICATES A DENDRON SUBSTITUTED INITIALLY WITH A FOCAL AROMATIC THIOL. FOR EACH
MOLECULE, FOUR IDENTICAL LIGANDS ARE ATTACHED TO THE IRON-SULFUR CORE (DENOTED BY A CIRCLED D)
CHAPTER 3
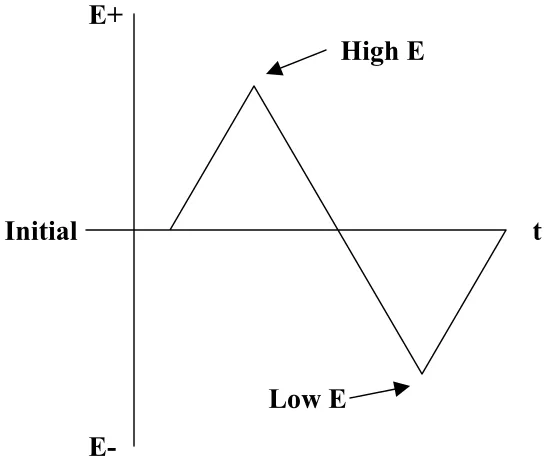
FIGURE 3.1 POTENTIAL WAVEFORM OF CYCLIC VOLTAMMETRY (POTENTIAL SWEEP)
FIGURE 3.2 CYCLIC VOLTAMMOGRAMS OF EACH EXTENDED (DOTTED) AND BACKFOLDED (SOLID) DENDRIMER ISOMER PAIR (1 mM
DENDRIMER, 100 mM TEAF IN DMF, SCAN RATE OF 50 mV/S)
FIGURE 3.3 PLOTS DISPLAYING LINEAR BEHAVIOR OF CURRENT VERSUS THE SQUARE ROOT OF SCAN RATE. THIS LINEAR RELATIONSHIP INDICATES A FREELY DIFFUSING SPECIES
FIGURE 3.4 WORKING CURVE SHOWING VARIATION OF PEAK POTENTIAL SEPARATION VERSUS Ψ
FIGURE 3.5 POTENTIAL WAVEFORM OF OSTERYOUNG SQUARE WAVE VOLTAMMETRY (POTENTIAL STEP)
FIGURE 3.6 POTENTIAL WAVEFORM OF CHRONOAMPEROMETRY (POTENTIAL PULSE)
FIGURE 3.7 ILLUSTRATION OF PFGSE 1H-NMR. THE ILLUSTRATION ON THE LEFT DEMONSTRATES THE MAGNETIC FIELD GRADIENT. THE EFFECT OF THIS GRADIENT IS SHOWN ON THE RIGHT AS
DIFFERENCES IN THE MAGNETIC SPIN VECTOR OF MOLECULES IN VARIOUS LOCATIONS WITHIN THE FIELD GRADIENT
FIGURE 3.8 CARTOON ILLUSTRATING THE ROLE OF CORE MOBILITY AND MOLECULAR SIZE ON THE EFFECTIVE ELECTRON-TRANSFER DISTANCE
FIGURE 3.10 A TYPICAL INVERSION RECOVERY EXPERIMENT SHOWING 1H-NMR SPECTRA AT MULTIPLE τD
FIGURE 3.11 BAR GRAPH INDICATING THE RELATIVE T1 VALUES OF TERMINAL AROMATIC PROTONS MEASURED FOR EACH
DENDRIMER (0.1 mM DMF SOLUTION)
FIGURE 3.12 CYCLIC VOLTAMMOGRAMS OF ALL DENDRIMERS AT MULTIPLE SCAN RATES
FIGURE 3.13 OSTERYOUNG SQUARE WAVE VOLTAMMOGRAMS OF ALL DENDRIMERS AT MULTIPLE SCAN RATES
FIGURE 3.14 NMR SPECTRA OF 2B,2B DENDRIMER AT MULTIPLE GRADIENT PULSE STRENGTHS (0.1 mM DENDRIMER IN D7-DMF). GRADIENT STRENGTHS (G/CM) FROM FRONT TO BACK 1.59, 7.95, 14.3, 20.7, 27.0, 33.4, 39.75, AND 47.2 (δ = 5 mS)
CHAPTER 4
FIGURE 4.1 CYCLIC VOLTAMMOGRAMS OF EACH CONSTITUTIONAL ISOMER AS A THIN FILM (DOTTED) AND IN SOLUTION (SOLID). (FILMS – 100 mM TEAPF6 IN 80:20 PROPYLENE GLYCOL/PROPYLENE CARBONATE, SCAN RATE OF 50 mV/S, VS. FC/FC+ EXTERNAL
STANDARD. SOLUTION – 1 mM DENDRIMER, 100 mM TEAF IN DMF, SCAN RATE OF 50 mV/S VS. FC/FC+ EXTERNAL STANDARD)
FIGURE 4.2 CYCLIC VOLTAMMOGRAMS OF EXTENDED (DOTTED) AND BACKFOLDED (SOLID) CONSTITUTIONAL ISOMERS AS THIN FILMS (100 mM TEAPF6 IN 80:20 PROPYLENE GLYCOL/PROPYLENE
CARBONATE, SCAN RATE OF 20 mV/S, VS. FC/FC+ EXTERNAL STANDARD)
FIGURE 4.3 SCHEMATIC OF THE THIN FILM DEPOSTION PROTOCAL. A 5 µL DROPLET WAS PRODUCED AT THE END OF A SYRINGE (LEFT), THE DROP WAS PLACED ON THE PT SURFACE (MIDDLE) AND ALLOWED TO DRY, UNDISTRUBED, TO CREATE A UNIFORM FILM ON THE PT BUTTON SURFACE (RIGHT)
FIGURE 4.4 CYCLIC VOLTAMMOGRAMS OF EXCESSIVELY THIN (TOP) AND THICK (BOTTOM) CONSTITUTIONAL ISOMER FILMS
FIGURE 4.5 CYCLIC VOLTAMMOGRAMS OF DENDRIMER THIN FILMS DEPOSITED FROM DENDRIMER/THF SOLUTION CONCENTRATIONS OF 500 µM, 1 mM AND 2 mM
FIGURE 4.6 CYCLIC VOLTAMMOGRAMS OF DENDRIMER THIN FILMS DEPOSITED FROM A 2 mM DENDRIMER/THF SOLUTION OF 1, 2 AND 5
– 5 µL DROP QUANTITIES
CHAPTER 5
FIGURE 5.1 STRUCTURE OF THE GENERATION 3, IRON-SULFUR
REPRESENT ADDITONAL ARMS IDENTICAL TO THE ONE SHOWN CLUSTER CORE DENDRIMER (G3 FLEX). THE CIRCLED – D’S
FIGURE 5.2 CYCLIC VOLTAMMOGRAMS OF DENDRIMER FILMS AS (A) THE COUNTER CATION WAS VARIED FROM TMA Æ TOA PF6 AND (B) THE COUNTER CAION WAS VARIED FROM TOA Æ TMA PF . SCAN RATE = 20 mV/S, PROPYLENE CARBONATE/100 mM SUPPORTING ELECTROLYTE, 10 mM AgNO3/100 mM TETRABUTYLAMMONIUM HEXAFLUOROPHOSPHATE IN MeCN REFERENCE ELECTRODE
6
FIGURE 5.3 VARIATION IN MEASURED REDOX POTENTIAL VERSUS RELATIVE DEBYE LENGTH FOR THREE OF THE FOUR
ELECTROLYTE SALTS STUDIED. THE SOLUBILITY OF TMAPF6 WAS INSUFFICIENT FOR THIS VARIATION
FIGURE 5.4 A TIME AVERAGED ILLUSTRATION OF THE IONIC
ATMOSPHERE AROUND A POSITIVELY (LEFT) AND NEGATIVELY (RIGHT) CHARGED ION
FIGURE 5.5 ILLUSTRATION OF THE RELATIONSHIP BETWEEN SIZE OF THE IONIC ATMOSPHERES OF A M+X- ELECTROLYTE AND LOW (LEFT) AND HIGH (RIGHT) IONIC STRENGTH
List of Tables
CHAPTER 3
TABLE 3-1. HETEROGENEOUS ELECTRON-TRANSFER RATE (kO), REDUCTION POTENTIAL (E1/2), AND TRANSFER COEFFICIENT (α) FOR THE ONE-ELECTRON REDOX COUPLE [Fe4S4(S-DEND)4]
2-/3-TABLE 3-2. DIFFUSION COEFFICIENTS, DO, OBTAINED FROM PULSED FIELD GRADIENT SPIN-ECHO NMR SPECTROSCOPY (PFGSE) AND CHRONOAMPEROMETRY (CA), AND THE CORRESPONDING STOKES-EINSTEIN RADIUS, RH, OF THREE IRON-SULFUR CORE
CONSTITUTIONAL ISOMER PAIRS
CHAPTER 4
TABLE 4-1. REDOX POTENTIAL FOR THE ONE-ELECTRON REDOX
COUPLE [Fe4S4(S-DEND)4]2-/3- IN SOLUTION AND DROP COATED AS A THIN FILM AND THE CORRESPONDING MOLECULAR WEIGHTS FOR IRON-SULFUR CORE G2 – G4 FLEX AND THREE PAIRS OF
CONSTITUTIONAL ISOMER DENDRIMERS
CHAPTER 5
TABLE 5-1. KINETIC PARAMETERS AND REDOX POTENTIAL IN VARIOUS HEXAFLUOROPHOPHATE SUPPORTING ELECTROLYTES OF G3 FLEX DENDRIMER THIN – FILMS
TABLE 5-2. KINETIC PARAMETERS OF G3 FLEX DENDRIMER THIN FILMS AFTER A 15 MINUTE PRE-SOAK IN VARIOUS
HEXAFLUOROPHOSPHATE SUPPORTING ELECTROLYTES
TABLE 5-3. DATA CALCULATIONS OF THIN FILM PROPERTEIS OF G3 FLEXIBLE IRON-SULFUR CORE DENDRIMERS DROP-COATED ON PLATINUM
64
75
100
120
122
List of Schemes
CHAPTER 2
SCHEME 2.1 SYNTHETIC SCHEME FOR 2,3 EXTENDED AND BACKFOLDED DENDRITIC ARCHITECTURES
SCHEME 2.2 SYNTHETIC SCHEME FOR 3,2 EXTENDED AND BACKFOLDED DENDRITIC ARCHITECTURES
SCHEME 2.3 SYNTHETIC SCHEME FOR 2,2 BACKFOLDED DENDRITIC ARCHITECTURE
SCHEME 2.4 SYNTHETIC SCHEME FOR THE
PROTECTION/DEPROTECTION OF THE DENDRITIC AROMATIC THIOL FOCAL UNITS
SCHEME 2.5 SYNTHETIC SCHEME FOR DENDRITIC LIGAND EXCHANGE WITH A Fe4S4 CLUSTER
35
36
36
37
Chapter 1
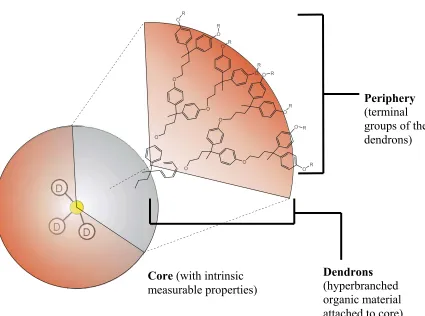
1.1 Origin of Dendrimers
Dendrimer chemistry began nearly two decades ago with the synthesis and characterization
of a family of highly branched macromolecules.1,2 The general dendrimer structure consisted
of an inner core molecule with hyperbranched polymeric structures, known as dendrons,
extending outward from the core. Each dendritic branch was terminated with a chosen
periphery structure (Figure 1.1). The ability to synthesize well-defined monodisperse
macromolecules using repetitive activate/couple growth cycles added a new dimension to
polymer chemistry.
Core (with intrinsic measurable properties)
Dendrons
(hyperbranched
attached to core)
Periphery
groups of the dendrons) (terminal
organic material
1.2 Dendrimer Synthesis: Convergent vs. Divergent
The development of new synthetic schemes enabled manipulation of the structure of
dendrimers. One scheme, the divergent strategy, consisted of an interactive synthesis
originating with the core and adding repeat units in an outward fashion (Figure 1.2).3 A
nearly monodisperse macromolecule with high molecular weights could be synthesized
through repetitive deprotection and coupling reactions.1 The disadvantage of the divergent
synthesis was the large excess of reagents required due to the large number of reactive sites.
To eliminate this problem, a convergent strategy for the synthesis of dendrimers was
developed. This strategy originates with the peripheral units and uses the same iterative
deprotection and coupling scheme to construct the dendritic branches inward (Figure 1.2).
Each coupling cycle designates a new layer of branch points referred to as a generation. The
dendritic arms are ultimately terminated with a linking unit that is used for core attachment.4
Monodispersity becomes a problem at high molecular weights therefore, the convergent
1.3 Dendrimer Encapsulation
It is known that the primary structure of the dendrons can be used to bias the conformation to
achieve general structures and shapes.6-15 These structures are governed by many factors
including solvation, sterics, hydrogen bonding, stereocenters, and benzyl substitution
patterns.16-27 Dendrimer structure is of significant interest due to the ability to encapsulate
core moieties at a sufficient size. The ability to effectively encapsulate the functional core is
interesting due to the possibility of revealing unique and potentially useful properties.3
1.3.1. Detection of Encapsulation: Photophysical Properties
The observation of photophysical changes were the first evidence of encapsulation behavior
of dendrimers.28-33 Hawker et al. coupled Frechet-type dendrons to a
4-(N-methylamino)-1-nitrobenzene core and observed an increase in the solvatochromic shift in the absorption
spectra as the dendrimer generation increased.28 The results indicated the larger generation
dendrimers effectively shielded solvent and created an intrinsic microenvironment around the
core. Additional techniques to further elucidate structure-property relationships of
dendrimers include fluorescence quenching,34-36 luminescence,37 and electrochemistry.36,38-46
1.3.2. Detection of Encapsulation: Electrochemical Properties
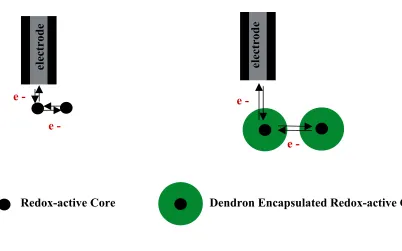
Dendrimers are attractive candidate molecules for studying electron transfer based solely on
the intrinsic ability of the dendrons (for a given molecular weight) to effectively shield the
core with organic material. The distance of electron travel and the material surrounding the
increasing the bulk material of the dendrons around the redox-active core, and/or 2) alter the
dendritic architecture to create a dynamic conformational change of the dendrimer. Both
strategies would result in an increase in the effective encapsulation and, subsequently, the
distance of electron transfer to the redox active core.
e - e -
e -
electrode electrode
e -
Redox-active Core Dendron Encapsulated Redox-active Core
Figure 1.3 Illustration of electron transfer between redox active molecules (left) and redox active molecules surrounded (encapsulated) by organic material (right).
Dendritic encapsulation has also been studied via changes in the kinetic and thermodynamic
properties of redox-active core dendrimers.6-8,16-19,36,37,39,41,46-64 Kinetic effects are manifested
as changes in the heterogeneous electron transfer rate, while thermodynamic changes become
apparent through shifts in redox potential. The attenuation of heterogeneous electron transfer
rates has been studied thoroughly, resulting in the further understanding of effective
encapsulation of redox active core moieties.6-8,16-19,36,39,41,46-61 In contrast, thermodynamics
potential shifts relate to structure-property relationships of redox-active dendrimers.
8,37,46,51,61-64
Research on electroactive dendrimers was introduced by Diederich and coworkers, who
studied the electron transfer kinetics of Zn-porphyrin core dendrimers encapsulated with
ethylene glycol terminated dendrons as a function of generation.46 As expected, the higher
generation molecules attenuated electron transfer more efficiently, displaying effective
encapsulation of the electroactive core. A decrease in electrochemical reversibility was also
seen by Frechet et al. with Zn-porphyrin cores surrounded by benzyl ether functionalities.36
In addition to porphyrin dendrimers, other types of dendrimers have displayed similar
electrochemical behavior during encapsulation studies.41
Kaifer, and matching work by Balzani, recently showed inconsistency with the general trend
of electron transfer attenuation with increasing generation.58,59 The molecules studied were
4,4’-bipyridinium core dendrimers containing Frechet-type benzyl-ether dendrons. Cyclic
voltammetry was used to measure the electron transfer kinetics using the Nicholson
analysis.65 It was shown that as dendrimer generation increased, the electron transfer kinetics
remained unchanged. This observation was contrary to all previous electronic studies of
redox-active dendrimer studies. However, a possible explanation is that due to the low
number of dendritic arms and therefore, low dendritic mass, effective encapsulation of the
bipyridine core dendrimers, including larger generation dendrons must be completed to fully
understand the observed results.
In addition to increasing mass, attempts have been made to elucidate structure-property
relationships by controlling the primary structure of the dendrons and studying the effects on
the overall conformation (shape) of the macromolecule.6-8,16-19,36,47,48,54-56 The introduction of
functional groups to achieve hydrogen bonding effects have been used previously to bias
dendron architecture.18,19 Synthetic attempts at restricting the intrinsic flexibility of these
macromolecules using chiral structures and acetylene units have been successful.17,47,48,54,55
Control of the primary architecture containing redox-active core moieties has been
studied.39,47,49-51,60,61 This research has shown the ability to change and control the primary
structure of the dendrimer. Biasing the dendritic architecture allows for the manipulation of
the molecule to further understand the factors governing the electronic property changes of
the redox-active core. Newkome and coworkers first synthesized asymmetrical Ruthenium
bis-terpyridine complexes39 and used cyclic voltammetry to probe the reversibility of the
redox reaction demonstrating an attenuation of the electron transfer rate. Chow et al. further
supported these finding on symmetric and asymmetric Ru-bis-terpy complexed dendrimers
from generations 1-3.49
The overall size of the dendrimer, the orientation of the branches, and the organic material of
coworkers explored the electrochemical behavior of asymmetric ferrocene core dendrimers.61
They found that encapsulation increased as generation increased, illustrated by a decrease in
the heterogeneous electron transfer rate to/from the electrode. They hypothesized that the
dendritic arm was flexible enough to backfold and effectively shield the ferrocene core.
Other encapsulation studies were completed with the asymmetric molecules including β
-cyclodextrin capping of the ferrocene units,51 and the use of charged peripheral groups to
orient the molecules at the electrode surface.60 Both experiments probed the manipulation of
dendritic material to effectively shield the core.
1.4 Dendrimer Applications
1.4.1. Electronic Materials
The ultimate goal of creating a molecule that could be synthesized to electronically trap and
accomplish electron trapping and potential unimolecular memory storage device applications.
Ideally, dendrimers tethered to a surface would encapsulate charge preventing “leaking” of
release this charge from molecule to molecule (Figure 1.4). This system could translate into
a binary representation where the charged molecules would represent ‘1s’ and a neutral
molecules, charge is intriguing. The encapsulating ability of dendrimers is desirable to ‘0s’.
Unimolecular storage would eliminate problems with mechanical scale down approaches,
which are approaching the lower limits of fabrication. Molecular devices would increase
memory storage capacities (per given area) well into the tera-byte realm versus the giga-byte
“0” “1”
Encapsulating Dendrimer Redox-active Molecule
“0” “1”
0 -1
0 -1
X
Figure 1.4 Illustration of the ideological use of dendrimers as charge trapping units to create a unimolecular binary memory system. Unencapsulated redox molecules (left) allow charge transfer between sites, scrambling information, while encapsulating dendrimers trap charge creating a binary system of 0’s (uncharged) and 1’s (charged).
1.4.2. Biological Models
Dendrimer interest spans not only within the area of molecular electronics, but also in the
biological model complexes where their function is governed by electron transfer.3,51,63 The
intrinsic ability of dendrimers to encapsulate and provide unique synthetic control creates an
ideal molecular model for determining the structure property relationships responsible for
tuning the electronic properties of biological molecules. Structural elements can be included
specifically to create structural facility that is unlike other attempts at creating protein mimics
and models.66-81 These attempts include mutant proteins68-73 and small molecule models.74-81
In all cases, problems surfaced when attempts were made to determine their
structure-property relationships. For mutants, determination of true structure and behavior can be
challenging,73 while small molecule models do not contain the steric protection required to
encapsulate and, ultimately, alter solvation of the functional moiety.76
1.5 Iron-Sulfur (Fe
4S
4) Core Dendrimers
Iron-sulfur clusters are particularly interesting redox centers due to their sensitivity to
changes of their surrounding environment. These changes can be detected by multiple
techniques including electrochemistry and nuclear magnetic resonance. Understanding how
dendritic structure and structural elements of the dendrimer affects the intrinsic, measurable
properties of the core, allows us to probe structure-property relationships of these molecules.
Elucidating structure-property relationships allows for the determination of the structural
Our interest in iron-sulfur clusters stem from their ubiquitous appearance in many biological
proteins. Iron-sulfur proteins are necessary for the execution of many life processes
including electron transfer across membranes82,83 and metabolic and enzymatic processes.84-86
Understanding the role of iron-sulfur clusters within biological proteins, may assist in
identifying the role of electron transfer as it relates to these life processes. More specifically,
understanding the structure-property relationships which govern/tune redox potential in
iron-sulfur proteins may provide insight of protein function and its’ relation to aging,87 disease,
87-89 and disorders.90.
Factors governing redox potential in iron-sulfur proteins have been discussed previously.
91-102 More specifically, considerable effort probing the effects of steric shielding,93,95-100
adjacent hydrogen bonding functionalities94,101,102 and solvation91,92 on redox behavior has
been attempted. However, these efforts via biological models/mimics, have encountered
many challenges.66,67,73,76,101,103
Holm et al. initiated the first studies of synthetic iron-sulfur cluster models to probe structural
effects governing redox potential.104-109 Their focus began with the successful synthesis of
iron-sulfur clusters (Fe4S4) outside of a protein environment. They discovered iron-sulfur
clusters were susceptible to facile ligand exchange with aromatic thiol terminated
ligands.74,110-112 Synthesis via ligand exchange created a rapid and selective synthetic route
for the construction of Fe4S4 molecular motifs. This facile synthesis was the impetus leading
have hypothesized that iron-sulfur core dendrimers contain the structural capability to control
the sulfur cluster microenvironment. To this end, our work focuses on the use of
iron-sulfur core dendrimers as a model to determine the factors governing the redox properties of
iron-sulfur clusters.
Our group has recently probed the effects of generation and dendritic architecture on the
kinetics of heterogeneous electron transfer.47 Iron-sulfur core dendrimers containing
“flexible” 4,4-bis(hydroxyphenyl)pentanol dendritic architectures, introduced by Frechet,
were directly compared to a “rigid” architecture containing phenylacetylene units (Figure
1.5, 1.6).52,53 Chronoamperometry, cyclic voltammetry, and Osteryoung square wave
voltammetry were used to find the diffusion coefficient, redox potential, and electron transfer
Figure 1.5 Generations 0-4 of “flexible” 4,4’-bis(hydroxyphenyl)pentanol repeat unit dendrimers (G0 - G4
Figure 1.6 Generations 0-4 of “rigid” 3,5-disubstituted phenylacetylene repeat unit dendrimers (G0 – G4
Rigid). Graphic from Gorman et al.47
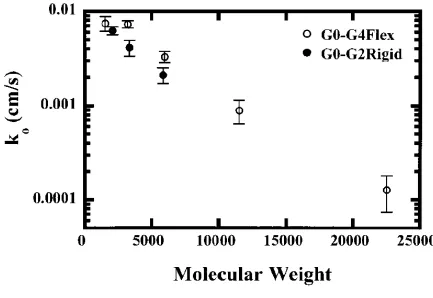
Results showed, as expected, that electron transfer rate attenuation occurred as the generation
increased for both types of dendrimers. Figure 1.7 displays electron transfer versus
molecular weight clearly indicating a greater attenuation for the phenylacetylene structure
the “rigid” architecture effectively encapsulated the core more efficiently than the “flexible”
architecture. This observation was intuitive since the phenylacetylene architecture allowed
for very little flexibility and was conformationally biased in which all branches were
extended, increasing the distance between the electrode surface and the iron-sulfur core.
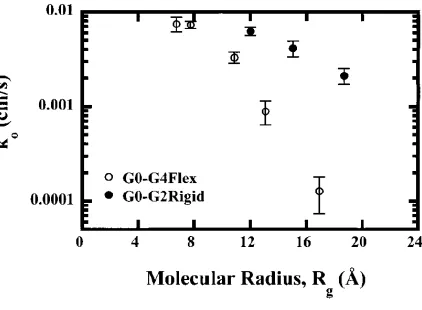
Conversely, the comparison of electron transfer versus the apparent radius of the dendrimer
revealed an interesting anomaly (Figure 1.8). The transfer of electrons through the “rigid”
architecture versus the “flexible” was, in fact, more facile as a function of size. It was
hypothesized that even though the cross-conjugated phenylacetylene branches were effective
in shielding the core from the electrode surface, the large number of π-bonds actually acted
as a pathway for through bond electron tunneling resulting in a nanowire effect. This
observation indicates that electron transfer is not only a distance phenomenon, but can be
Figure 1.7 Heterogeneous electron transfer rates for the “flexible” and “rigid” dendrimers plotted as a function
Figure 1.8 Heterogeneous electron transfer rates for the “flexible” and “rigid” dendrimers plotted as a function
of the molecular radius of gyration. Graphic from Gorman et al.47
In addition, to heterogeneous electron transfer kinetics, the thermodynamic redox potentials
have also been noted in the literature.8,37,46,51,61-64 Gorman and Smith hypothesized that redox
potential shifts were controlled by the microenvironment around the redox active core.63 The
microenvironment was defined by the ability of the dendritic material to stabilize/destabilize
the charge associated with the particular redox core. Electronically, changes in the
microenvironment are denoted as relative shifts in the HOMO/LUMO gap resulting in a
change in the energy required to add/remove an electron into/out of the energy levels. In
this hypothesis, however, further research must be done to truly understand how the
HOMO/LUMO gap of the redox-active core can be controlled directly by dendritic structure.
1.6 Perspectives of Dissertation Research
A synthetic scheme has recently been reported using substitution patterns around phenyl
rings to manipulate the dendron structure resulting in a backfolded architecture.16 In this
work, an attempt was made to bias dendrimer conformation into a more backfolded
architecture by way of benzyl ring substitution patterns resulting in a more shape persistent
structure. Chapter 2 reports the synthesis of three constitutional isomer pair dendrimers to
compare extended and backfolded architectures that are independent of molecular weight.
This bias of dendritic architecture was expected to increase encapsulation around an
environmentally sensitive iron-sulfur core, resulting in heterogeneous electron transfer rate
attenuation (Chapter 3). The use of electrochemical techniques on dendrimers containing
redox-active cores have shown to be useful in the attempt to elucidate structure-property
relationships.8,42,47 In addition, a change in the dynamic conformation of the dendrimers was
probed using nuclear magnetic techniques (Chapter 3). The use of NMR to probe dendrimer
structure has been employed previously.47,113
To further understand structure-property relationships governing the electronic behavior of
iron-sulfur clusters, studies were carried out on iron-sulfur core dendrimer thin films.
role in governing the electronic properties of redox-active molecules.114-117 In many cases,
the protein creates an environment where structural material governs the electronic properties
of iron-sulfur core dendrimers due to the exclusion of solvent.118 Therefore, thin films of
iron-sulfur core dendrimers were utilized to create a solvent excluded environment where the
electronic properties and structure-property relationships of iron-sulfur clusters could be
probed (Chapter 4 & 5). In these experiments, dendritic material and counterion migration
1.7 References
1. Tomalia, D. A.; Baker, A.; Dewald, J.; Hall, M.; Kallos, G.; Martin, S.; Roeck, J.; Ryder, J. and Smith, P. Polymer Journal 198517, p. 117-132.
2. Newkome, G. R.; Yao, Z.-Q.; Baker, G. R. and Gupta, V. K. Journal of Organic
Chemistry 198550, p. 2003-2004.
3. Frechet, J. M. J., Hecht, S. Angewandte Chemie-International Edition 200140, p. 74-91.
4. Frechet, J. M. J. and Hawker, C. Abstracts of Papers of the American Chemical Society
1990199, p. 316-ORGN.
5. Frechet, J. M. J. and Grayson, S. K. Chemical Reviews 2001101, p. 3819-3867.
6. Ballauff, M. Dendrimers Iii: Design, Dimension, Function 2001212, p. 177-194.
7. Wyatt, S. R.; Hu, Q. S.; Yan, X. L.; Bare, W. D. and Pu, L. Macromolecules 200134, p. 7983-7988.
8. Stone, D. L., Smith, D.K., McGrail, P.T. Journal of the American Chemical Society
2002124, p. 856-864.
9. Percec, V. Pure and Applied Chemistry 199567, p. 2031-2038.
10. Percec, V., Cho, W.D., Moller, M., Prokhorova, S., Ungar, G., Yeardley, D.J.P Journal
of the American Chemical Society 2000122, p. 4249-4250.
11. Percec, V.; Chu, P.; Ungar, G. and Zhou, J. Journal of the American Chemical Society
1995117, p. 11441-11454.
12. Percec, V.; Ahn, C.-H.; Ungar, G.; Yeardley, D. J. P.; Möller, M. and Sheiko, S. S.
13. Percec, V.; Cho, W. D.; and Ungar, G. Abstracts of Papers of the American Chemical
Society 1997214, p. 141-PMSE.
14. Percec, V.; Cho, W. D.; Mosier, P. E.; Ungar, G. and Yeardley, D. J. P. Journal of the
American Chemical Society 1998120, p. 11061-11070.
15. Percec, V.; Cho, W. D.; Ungar, G. and Yeardley, D. J. P. Journal of the American
Chemical Society 2001123, p. 1302-1315.
16. Peerlings, H. W. I., Trimbach, D. C., and Meijer, E. W. Chemical Communications
1998 p. 497-498.
17. Recker, J.; Tomcik, D. J. and Parquette, J. R. Journal of the American Chemical Society
2000122, p. 10298-10307.
18. Gandhi, P.; Huang, B. H.; Gallucci, J. C. and Parquette, J. R. Organic Letters 20013, p. 3129-3132.
19. Huang, B. H. and Parquette, J. R. Journal of the American Chemical Society 2001123, p. 2689-2690.
20. Lescanec, R. L. and Muthukumar, M. Macromolecules 199023, p. 2280-2288.
21. Murat, M. and Grest, G. S. Macromolecules 199629, p. 1278-1285.
22. Mansfield, M. L. and Klushin, L. I. Journal of Physical Chemistry 199296, p. 3994-3998.
23. Mansfield, M. L. and Klushin, L. I. Macromolecules 199326, p. 4262-4268.
24. Carl, W. Journal of the Chemical Society, Faraday Transactions 199692, p. 4151-4154.
26. Chen, Z. Y. and Cui, S.-M. Macromolecules 199629, p. 7943-7952.
27. Boris, D. and Rubinstein, M. Macromolecules 199629, p. 7251-7260.
28. Hawker, C. J., Wooley, K.L., Frechet, J.M.J Journal of the American Chemical Society
1993115, p. 4375-4376.
29. Smith, D. K. and Muller, L. Chemical Communications 1999 p. 1915-1916.
30. Balzani, V.; Vogtle, F.; Plevoets, M.; Nieger, M.; Azzellini, G. C.; Credi, A.; De Cola, L.; De Marchis, V. and Venturi, M. Journal of the American Chemical Society 1999
121, p. 6290-6298.
31. Fréchet, J. M. J.; Adronov, A. and Gilat, S. L. Angewandte Chemie-International
Edition 199938, p. 1422-1427.
32. Fréchet, J. M. J.; Adronov, A.; Gilat, S. L.; Ohta, K.; Neuwahl, F. V. R. and Fleming, G.
R. Journal of the American Chemical Society 2000122, p. 1175-1185.
33. Fréchet, J. M. J.; Adronov, A. and Malenfant, P. R. L. Chemistry of Materials 200012, p. 1463-1472.
34. Aida, T.; Tomioka, N.; Takasu, D. and Takahashi, T. Angewandte Chemie-International
Edition 1998110, p. 1611-1614.
35. Aida, T.; Jin, R. H. and Inoue, S. Journal of the Chemical Society, Chemical
Communications 1993 p. 1260-1262.
36. Abruna, H. D.; Pollak, K. W.; Leon, J. W.; Frechet, J. M. J. and Maskus, M. Chemistry
of Materials 199810, p. 30-38.
37. Fréchet, J. M. J. and Kawa, M. Chemistry of Materials 199810, p. 286-296.
38. Chow, H. F.; Chan, I. Y. K.; Chan, D. T. W. and Kwok, R. W. M. Chemistry-a
39. Newkome, G. R.; Güther, R.; Moorefield, C. N.; Cardullo, F.; Echegoyen, L.; Pérez-Cordero, E. and Luftmann, H. Angewandte Chemie-International Edition 199534, p. 2023-2026.
40. Chen, K.-Y. and Gorman, C. B. Journal of Organic Chemistry 199661, p. 9229-9235.
41. Gorman, C. B.; Parkhurst, B. L.; Chen, K.-Y. and Su, W. Y. Journal of the American
Chemical Society 1997119, p. 1141-1142.
42. Gorman, C. B. Advanced Functional Materials 19979, p. 1117-1119.
43. Diederich, F.; Weyermann, J. P.; Gisselbrecht, P.; Boudon, C. and Gross, M.
Angewandte Chemie-International Edition 199938, p. 3215-3219.
44. Dandliker, P. J.; Diederich, F.; Zingg, A.; Gisselbrecht, J. P.; Gross, M.; Louati, A. and Sanford, E. Helvetica Chimica Acta 199780, p. 1773-1801.
45. Dandliker, P. J.; Diederich, F.; Gisselbrecht, J.-P.; Louati, A. and Gross, M. Angewandte
Chemie-International Edition 199534, p. 2725-2728.
46. Dandliker, P. J.; Diederich, F.; Gross, M.; Knobler, C. B.; Louati, A. and Sanford, E. M.
Angewandte Chemie-International Edition 199433, p. 1739-1741.
47. Gorman, C. B., Smith, J. C., Hager, M. W., Parkhurst, B. L., Sierzputowska-Gracz, H., Haney, C. A. Journal of the American Chemical Society 1999121, p. 9958-9966.
48. Rosini, C.; Peerlings, H. W. I.; Superchi, S. and Meijer, E. W. European Journal of
Organic Chemistry 2000 p. 61-71.
49. Chow, H. F.; Chan, I. Y. K.; Fung, P. S.; Mong, T. K. K. and Nongrum, M. F.
Tetrahedron 200157, p. 1565-1572.
50. Kaifer, A. E.; Cardona, C. M. and McCarley, T. D. Journal of Organic Chemistry 2000
51. Kaifer, A. E.; Cardona, C. M. and Mendoza, S. Chemical Society Reviews 200029, p. 37-42.
52. Xu, Z.; Kahr, M.; Walker, K. L.; Wilkins, C. L. and Moore, J. S. Journal of the
American Chemical Society 1994116, p. 4537-4550.
53. Moore, J. S. and Xu, Z. F. Macromolecules 199124, p. 5893-5894.
54. Gorman, C. B. and Smith, J. C. Polymer 200041, p. 675-683.
55. Pesak, D. J. and Moore, J. S. Angewandte Chemie-International Edition in English
199736, p. 1636-1639.
56. Scherrenberg, R.; Coussens, B.; van Vliet, P.; Edouard, G.; Brackman, J.; de Brabander, E. and Mortensen, K. Macromolecules 199831, p. 456-461.
57. Cameron, C. S. and Gorman, C. B. Advanced Functional Materials 200212, p. 17-20.
58. Balzani, V.; Vicinelli, V.; Maestri, M.; Muller, W. M.; Muller, U.; Hahn, U.; Osswald, F. and Vogtle, F. New Journal of Chemistry 200125, p. 989-993.
59. Kaifer, A. E.; Toba, R.; Quintela, J. M.; Peinador, C. and Roman, E. Chemical
Communications 2001 p. 857-858.
60. Kaifer, A. E.; Wang, Y. and Cardona, C. M. Journal of the American Chemical Society
1999121, p. 9756-9757.
61. Kaifer, A. E. and Cardona, C. M. Journal of the American Chemical Society 1998120, p. 4023-4024.
62. Weyermann, P.; Diederich, F.; Gisselbrecht, J. P.; Boudon, C. and Gross, M. Helvetica
Chimica Acta 200285, p. 571-598.
64. Kaifer, A. E. and Ong, W. Journal of the American Chemical Society Communications
2002124, p. 9358-9359.
65. Nicholson, R. S. Analytical Chemistry 196537, p. 1351-1355.
66. Cammack, R. Advances in Inorganic Chemistry 199238, p. 281-322.
67. Fukuyama, K., Ferredoxins containing one [4Fe-4S] center, in Handbook of
Metalloproteins, A. Messerschmidt, R. Huber, T. Poulos, and K. Weighardt, Editors.
2001. Wiley: Chichester ; New York. p. 543-552.
68. Shen, B. H.; Jollie, D. R.; Stout, C. D.; Diller, T. C.; Armstrong, F. A.; Gorst, C. M.; La Mar, G. N.; Stephens, P. J. and Burgess, B. K. Journal of Biological Chemistry 1994
269, p. 8564-8575.
69. Chen, K. S.; Tilley, G. J.; Sridhar, V.; Prasad, G. S.; Stout, C. D.; Armstrong, F. A. and Burgess, B. K. Journal of Biological Chemistry 1999274, p. 36479-36487.
70. Low, D. W. and Hill, M. G. Journal of the American Chemical Society 2000122, p. 11039-11040.
71. Babini, E.; Bertini, I.; Borsari, M.; Capozzi, F.; Luchinat, C.; Zhang, X. Y.; Moura, G. L. C.; Kurnikov, I. V.; Beratan, D. N.; Ponce, A.; Di Bilio, A. J.; Winkler, J. R. and Gray, H. B. Journal of the American Chemical Society 2000122, p. 4532-4533.
72. Armstrong, F. A.; Butt, J. N.; Govindaraju, K.; McGinnis, J.; Powls, R. and Sykes, A. G.
Inorganic Chemistry 199029, p. 4858-4862.
73. Martin, A. E.; Burgess, B. K.; Stout, C. D.; Cash, V. L.; Dean, D. R.; Jensen, G. M. and Stephens, P. J. Proceedings of the National Academy of Sciences of the United States of
America 199087, p. 598-602.
74. Que Jr., L.; Holm, R. H.; Bobrik, M. A. and Ibers, J. A. Journal of the American
75. Laplaza, C. E. and Holm, R. H. Journal of the American Chemical Society 2001123, p. 10255-10264.
76. Holm, R. H. Accounts of Chemical Research 197710, p. 427-434.
77. Mulholland, S. E.; Gibney, B. R.; Rabanal, F. and Dutton, P. L. Journal of the American
Chemical Society 1998120, p. 10296-10302.
78. Mulholland, S. E.; Gibney, B. R.; Rabanal, F. and Dutton, P. L. Biochemistry 199938, p. 10442-10448.
79. Ueyama, N.; Terakawa, T.; Nakata, M. and Nakamura, A. Journal of the American
Chemical Society 1983105, p. 7098-102.
80. Nakamura, A. and Ueyama, N. American Chemical Society Symposium Series 1988
372, p. 292-301.
81. Nakamura, A. and Ueyama, N. Advanced Inorganic Chemistry 198933, p. 39-67.
82. Jormakka, M.; Tornroth, S.; Byrne, B. and Iwata, S. Science 2002295, p. 1862-1868.
83. Johnson, M. K., Iron-Sulfur Proteins, in Encyclopedia of Inorganic Chemistry, R.B. King, Editor. 1994, Wiley: Chichester ; New York. p. 1896-1915.
84. Broderick, J. B.; Duderstadt, R. E.; Fernandez, D. C.; Wojtuszewski, K.; Henshaw, T. F. and Johnson, M. K. Journal of the American Chemical Society 1997119, p. 7396-7397.
85. Daley, C. J. A. and Holm, R. H. Inorganic Chemistry 200140, p. 2785-2793.
86. Busby, R. W.; Schelvis, J. P. M.; Yu, D. S.; Babcock, G. T. and Marletta, M. A. Journal
of the American Chemical Society 1999121, p. 4706-4707.
87. Atamna, H.; Walter, P. B. and Ames, B. N. Archives of Biochemistry and Biophysics
88. Schapira, A. H. V. Biochimica et Biophysica Acta 19981364, p. 261-270.
89. Ebadi, M.; Govitrapong, P.; Sharma, S.; Muralikrishnan, D.; Shavali, S.; Pellett, L.; Schafer, R.; Albano, C. and Eken, J. Biological Signals and Receptors 200110, p. 224-253.
90. Puccio, H. and Koening, M. Clinical Neuroscience Research 20011, p. 127-133.
91. Soriano, A.; Li, D. W.; Bian, S. M.; Agarwal, A. and Cowan, J. A. Biochemistry 1996
35, p. 12479-12486.
92. Capozzi, F.; Ciurli, S. and Luchinat, C. Structure and Bonding: Metal Sites in Proteins and Models. Vol. 90. 1998. 127-160.
93. Boll, M.; Fuchs, G.; Tilley, G.; Armstrong, F. A. and Lowe, D. J. Biochemistry 2000
39, p. 4929-4938.
94. Sheridan, R. P.; Allen, L. C. and Carter, C. W. Journal of Biological Chemistry 1981
256, p. 5052-5057.
95. Fukuyama, K.; Matsubara, H.; Tsukihara, T. and Katsube, Y. Journal of Molecular
Biology 1989210, p. 383-398.
96. Banci, L.; Bertini, I.; Ciurli, S.; Ferretti, S.; Luchinat, C. and Piccioli, M. Biochemistry
199332, p. 9387-9397.
97. Davy, S. L.; Osborne, M. J. and Moore, G. R. Journal of Molecular Biology 1998277, p. 683-706.
98. Sticht, H.; Wildegger, G.; Bentrop, D.; Darimont, B.; Sterner, R. and Rosch, P.
European Journal of Biochemistry 1996237, p. 726-735.
100. Wang, P. L.; Donaire, A.; Zhou, Z. H.; Adams, M. W. W. and La Mar, G. N.
Biochemistry 199635, p. 11319-11328.
101. Babini, E.; Bertini, I.; Borsari, M.; Capozzi, F.; Dikiy, A.; Eltis, L. D. and Luchinat,
C. Journal of the American Chemical Society 1996118, p. 75-80.
102. Glaser, T.; Bertini, I.; Moura, J. J. G.; Hedman, B.; Hodgson, K. O. and Solomon, E.
I. Journal of the American Chemical Society 2001123, p. 4859-4860.
103. Agarwal, A.; Li, D. W. and Cowan, J. A. Journal of the American Chemical Society
1996118, p. 927-928.
104. Herskovitz, T.; Averill, B. A.; Holm, R. H.; Ibers, J. A.; Phillips, W. D. and Weiher, J. F. Proceedings of the National Academy of Sciences 197269, p. 2437-2441.
105. Holm, R. H.; Averill, B. A.; Herskovitz, T.; Frankel, R. B.; Gray, H. B.; Siiman, O. and Grunthaner, F. J. Journal of the American Chemical Society 197496, p. 2644-6.
106. Averill, B. A.; Herskovitz, T.; Holm, R. H. and Ibers, J. A. Journal of the American
Chemical Society 197395, p. 3523-3534.
107. Holm, R. H.; Phillips, W. D.; Averill, B. A.; Mayerle, J. J. and Herskovitz, T.
Journal of the American Chemical Society 197496, p. 2109-2117.
108. DePamphilis, B. V.; Averill, B. A.; Herskovitz, T.; Que Jr., L. and Holm, R. H.
Journal of the American Chemical Society 197496, p. 4159-4167.
109. Reynolds, J. G.; Laskowski, E. J. and Holm, R. H. Journal of the American Chemical
Society 1978100, p. 5315-5322.
110. Johnson, R. W. and Holm, R. H. Journal of the American Chemical Society 1978
100, p. 5338-5344.
112. Bobrik, M. A.; Que Jr., L. and Holm, R. H. Journal of the American Chemical
Society 197496, p. 285-287.
113. Gorman, C. B.; Hager, M. W.; Parkhurst, B. L. and Smith, J. C. Macromolecules
199831, p. 815-822.
114. Anderson, J. L.; Bowden, E. F. and Pickup, P. G. Analytical Chemistry 199668, p. R379-R444.
115. Clark, R. A. and Bowden, E. F. Langmuir 199713, p. 559-565.
116. El Kasmi, A.; Wallace, J. M.; Bowden, E. F.; Binet, S. M. and Linderman, R. J.
Journal of the American Chemical Society 1998120, p. 225-226.
117. Armstrong, F. A.; Heering, H. A. and Hirst, J. Chemical Society Reviews 199726, p. 169-179.
Chapter 2
Synthesis and Characterization of Constitutional Isomers of
Benzyl-Ether Iron-Sulfur Core Dendrimers
The synthetic efforts of Rakesh Sachdeva, Qun Li, and Zemin Li, as well as Randall J. Petrie and his Matrix Assisted Laser Desorption Ionization expertise should not go unmentioned and is gratefully acknowledged here.
2.1 Introduction
A decisive way to probe the influence of structure on encapsulation via conformation is
through the study of constitutional isomers. The synthesis1,2 and study of dendrimer isomers
has had some precedent including the study of linear versus hyperbranched structures,3,4
supramolecular organization of different dendrimer isomers,5 stereoisomers in dendrimers
(e.g. cis vs. trans azobenzene or stilbene linkages in dendrimers)6-13 and isomeric
metallodendrimers.14 Recently, the relative rates of photoinduced, intramolecular electron
transfer of constitutional isomers of a triphenylamine core dendrimer substituted with a
peryleneimide acceptor at the periphery were probed.15
Ideal dendrimer isomers differ only in their primary structure. Changes in the primary
structure of the dendrimer can result in a change in its’ conformation (e.g. the disposition of
the arms around the core and the relative degree to which the core is buried within the
dendrimer). This change in conformation could be reflected in a change in the measured
degree of encapsulation of the core. For example, in the case of an electroactive core (such
as is discussed here), a change in the kinetics and/or thermodynamics of electron transfer
to/from that core is expected.
Several notable efforts have elucidated structure-property relationships by changing the
primary structure of the dendrons to bias dendrimer conformation.1,16-22 Conformation can
internal hydrogen bonding sites,24-27 and steric factors including chiral moieties within the
primary structure.6,28-36
S O O O O O O O O O O O O O O S O O O O O O O O O S
O O O
O O O O O O O S O O O O O O O O O S O O O O O O O O O S O Fe S S S Fe Fe Fe S
O
DO
DO
DO
D 2,3 Backfolded (2B, 3)3,2 Extended (3,2)
2,2 Extended (2,2)
3,2 Backfolded (3,2B)
2,2 Backfolded (2B,2B)
2,3 Extended (2,3)
Figure 2.1. Structures of the constitutional isomeric dendrimer pairs. Each dendrimer has the form
(nBu4N)2{Fe4S4D4}, where D indicates a dendron substituted initially with a focal aromatic thiol. For each
molecule, four identical ligands are attached to the iron-sulfur core (denoted by a circled D).
We and others29,30,37-43 have hypothesized that primary structural elements that create
around the core moiety of the dendrimer. This behavior should lead to more effective
encapsulation. To probe this hypothesis experimentally, three pairs of constitutional
isomeric dendrimers containing benzyl-ether repeat units (Figure 2.1) were synthesized and
studied. These molecules allow the direct comparison of extended and backfolded
architecture independent of the molecular weight.
2.2. Results and Discussion
2.2.1. General Synthetic Methods
Each dendrimer structure synthesized consisted of benzyl ether-based repeat units previously
Scheme 2.1 Synthetic scheme for 2,3 extended and backfolded dendritic architectures. O O O Cl O O O
O O O O
O X O O O O O O O O X
X=CO2CH3
X=CH2Cl
X=CH2OH
X=CH2OH
X=CH2Br
CH2OH
HO OH
K2CO3
Acetone reflux/72hrs
DCM
0o/2hrs
70% 70%
CO2CH3
OH HO
LiAlH4, THF
0o/2hrs
82%
DCM 0o/2hrs
76%
CBr4, PPh3,
89%
CCl4, PPh3
Scheme 2.2 Synthetic scheme for 3,2 extended and backfolded dendritic architectures.
X=CO2CH3 X=CH2OH
X=CH2Cl
X=CH2Br
X O O O O O O O O O X O O O O O O O O O O O O O Cl Br 93% 86%
K2CO3
Acetone reflux/72hrs
CO2CH3
HO
OHOH
LiAlH4, THF
0o/2hrs
90%
87%
SOCl2, DCM
Proton Sponge
DCM 0o/2hrs
63%
CBr4, PPh3
70%
0o/2hrs
7 8 9,13 10 14 11 12
Scheme 2.3 Synthetic scheme for 2,2 backfolded dendritic architecture.
X=CO2CH3
X=CH2OH X=CH2Br
O O
Br
K2CO3 Acetone reflux/72 hrs
LiAlH4, THF 0o/2 hrs
Et2O/DCM 0o/30 min
PBr3 11 O O O O O O X HO OH
O OCH3
Scheme 2.4 Synthetic scheme for the protection/deprotection of the dendritic aromatic thiol focal units.
3,2-Cl
3,2B-Br
2,3-Cl
2B,3-Br
O
OH S
K2CO3 Acetone 72 hrs, reflux
X =
O
O S X =
AgNO3/aq. THF NaSH/DCM
90 min, rt O S 3
10 14
19 2,3-X 60%
20 2B,3-X 90%
21 3,2-X 75%
22 3,2B-X 84%
23 2,2-X 67%
24 2B,2B 49%
37
2,2-Br
18
2B,2B-Br
17
25 2,3-X 93%
26 2B,3-X 94%
27 3,2-X 88%
28 3,2B-X 91%
29 2,2-X 93%
30 2B,2B 87%
6
H
Scheme 2.5 Synthetic scheme for dendritic ligand exchange with a Fe4S4 cluster.
O SH
X = + DMF, 1hr
N2 Fe S S S Fe
FeS Fe
O
DO
DO
DX =
O
D2B,3-X 25 27 28 2,2-X
30 2B,2B-X
29 26 3,2-X 3,2B-X 2,3-X
{Fe4S4[SC(CH3)3]4}[N((CH2)3CH3)4]2
31 2,3-X 80% 32 2B,3-X 80% 33 3,2-X 90% 34 3,2B-X 86% 35 2,2-X 74% 36 2B,2B 62%
Each dendrimer was synthesized using a convergent activate/couple approach (Schemes 2.1 –
2.5). The extended and backfolded designations refer to the aromatic substitution patterns of
the benzyl ether groups. Isomers containing 3,5-di-substituted linkages are designated as
It is important to note that significant care had to be taken with all benzyl ether dendrons due
to their susceptibility to acid cleavage of the benzyl ether groups as detailed previously.21,39
Reaction and purification steps had to be carried out in neutral or slightly basic conditions for
these particular compounds.
2.2.1.1. General Method for Halogenation
The halogenation synthetic steps were substitution reactions converting a benzyl alcohol to a
benzyl halide. This reaction was required to activate the dendritic branch for further
coupling with phenol functionalities resulting in subsequent generation increases. Further,
the halogenation of G2 dendrons acts as an activation step prior to coupling with the
dendritic focal unit.
Multiple halogenation schemes were attempted for each dendron. In many cases, however,
only one of the selected halogenation schemes resulted in the desired product. This result
was primarily due to particular dendrons increased susceptibility to hydrolysis from acidic
side products. Solutions included introducing a weak base to neutralize acidic side products
and using different halides (Cl, Br) to exploit differences in reactivity.
Further, the halogenated compounds were carried on immediately without purification. In
multiple instances, halogenated compounds that were allowed to sit for extended periods of
time, degraded rapidly. The degradation was apparent by 1H-NMR and immediately
visualized by a significant color and texture change from a white powder to a dark, red/black
colorless to a deep red. During purification, this band remained stationary and the overall
yield was significantly reduced. All halogenated intermediates were thus carried on without
purification.
General Method for Chlorination (SOCl2)
To a solution of the alcohol (1 equiv) and proton sponge (1.5 equiv) dissolved in methylene
chloride and cooled in an ice bath, was added thionyl chloride (1.1 equiv) dropwise while
stirring. The mixture was allowed to warm to room temperature under continuous stirring.
After 2h, a color change was observed and the mixture was quenched with water. The
organics were extracted with methylene chloride three times. The combined organic extracts
were washed with brine, dried over anhydrous sodium sulfate, filtered and concentrated.
General Method for Chlorination (CCl4/PPh3)
To a solution of the alcohol (1.0 equiv) at 0oC, tri-phenyl phosphine (1.3 equiv) and carbon
tetra-chloride (3.5 equiv) at 0oC was cannulated dropwise while stirring. The temperature
was allowed to rise to ambient over 2h. The reaction was quenched with water, the organics
were extracted with DCM, washed with saturated NaCl and dried using MgSO4. The mixture
was filtered and concentrated.
General Method for Bromination (CBr4/PPh3)
To a solution of the alcohol (1.0 equiv) and tri-phenyl phosphine (1.1 equiv) in THF or DCM
equiv) in THF or DCM while stirring. The temperature was allowed to rise to ambient over
2h. The reaction was quenched with water and stirred for 2 hours. The organics were
extracted with DCM, washed with saturated NaCl and dried using Na2SO4 or MgSO4. The
mixture was filtered and concentrated.
General Method for Bromination (PBr3)
To a solution of the alcohol (2.7 equiv) in Et2O or THF cooled in an ice bath, was added a
solution of PBr3 (1.0 equiv) in Et2O or THF, dropwise via cannulation while stirring. The
temperature was kept at 0oC for 1 hour. The reaction was warmed to room temperature and
quenched with water. The organics were extracted with ethyl acetate and washed with
saturated sodium bicarbonate and dried using Na2SO4. The mixture was filtered and
concentrated.
2.2.1.2 General Method for Coupling Reaction
To a solution of the phenol derivative (1.0 equiv), dry acetone (50-60 equiv), potassium
carbonate (1.1 equiv per OH in phenol), and 18-crown-6 (0.01-0.02 equiv) was added a
benzyl halide derivative (1.1-1.2 equiv per OH in phenol). The mixture was then refluxed for
2-3 days under nitrogen with vigorous stirring. The mixture was cooled and concentrated to
half the volume. Water was added to dissolve the salts and the organics extracted into ethyl
acetate, methylene chloride, or ether. The organic extract was washed with brine or NaHCO3
under reduced pressure gave the crude product. The individual purification techniques are
shown for each compound in Section 2.3. Experimental/Compound Data.
2.2.1.3. General Method for Reduction
The ester (1.0 equiv) was cannulated into a solution of LiAlH4 (1.3 equiv) in dry THF (21
equiv) cooled in an ice-water bath stirring continuously for 2 hours. The reaction was
quenched with H2O, 15% NaOH, or aqueous NH4Cl. This was followed by addition of 6M
HCl to dissolve the salts. After the reaction was complete (few hours for extended and
overnight for backfolded), the reaction was carefully worked up with water and the solids
filtered by passing through a bed of Celite. The organics were extracted into ethyl acetate or
DCM. The organic layer was dried over anhydrous Na2SO4, filtered, concentrated, and
placed under vacuum overnight. The crude alcohol was purified as shown in Section 2.3.
Experimental/Compound Data.
2.2.1.4. General Method for Deprotection
Tetrahydropyran (THP) protected thiol 37 (Experimental/Compound Data Section 2.3)(1.0
equiv) was dissolved in tetrahydrofuran. Excess of silver nitrate was added. Water was then
added to the mixture until bright yellow precipitate forms and the solution was stirred for 20
min. The mixture was diluted ten-fold with DCM. NaSH (solid, 10 equiv) was added and
stirred vigorously for thirty minutes. The result was a black precipitate, which was removed
by filtration. The precipitate was washed twice with DCM. The aqueous and organic phases