The
Oxidation
of
Alcohols
to
Aldehydes
or
Ketones
HIGH OXIDATION STATE RUTHENIUM COMPOUNDS AS CATALYSTS
By Holger
B.
Friedrich
School of Pure &- Applied Chemistry, University of Natal, South Africa
T h e oxidations of alcohols to aldehydes and ketones are vital reurliuns in
synthetic organic chemistry, and high selectivity and mild conditions are important prerequisites for ease of product work-up and lower cost. Currentl-y, m a n y of the best oxidants for these conversions contain high valent ruthe- nium, with ruthenium acting as a catalyst for these reactions. I t is important to have detailed knowledge and understanding of the mechanisms of the oxidation reactions and the factors that injluence them, as only by completely understanding how these proresses work will it be possible to design better or optimal catalysts. I n this paper, the more viable oxidants currently available are reported and some investigations into the mechanisms of the reartions and factors affecting them are discussed.
As the catalytic conversions of primary alco- hols into aldehydes and of secondary alcohols into ketones are essential for the preparation of many key synthetic intermediates in organic chemistry, there is currently much activity being undertaken in the search for catalytic systems which can perform these operations.
oxidant
oxidant
RCHiOH A RCHO ( 0
RCH(0H)R’ A RC(0)R’ (ii)
In particular, the reaction shown in Equation (i) does not usually stop at the formation of the aldehyde and over-oxidation to the carboxylic acid often occurs.
H 3 0
RCHO
,
RCOOH (iii)Historically, various oxidative systems have been available for these reactions, including chromium(V1) 0x0 compounds, copper chromites (often used industrially for bulk prepa- rations), vanadium pentoxide, PtO,, RuCl,/Cu(II) (1, 2) and the Swern oxidation ( 3 ) . However, there are various drawbacks to using these reagents; for instance, they are either rather aggressive, need relatively high or low temperatures, give unwanted and difficult to
remove by-products, are difficult to separate from the product after the reaction is over, require acidic conditions which limit the choice of substrate, or, they do not give complete con- versions in all applications. Thus, there is clearly a need to prepare milder and more selective oxi- dants with higher rates of conversion. Several oxidative systems which at least partially fit
the above criteria have recently been reported, and many of these contain or are based on ruthenium.
The chemistry of high-oxidation-state ruthe- nium is less well developed than that of, for example, osmium (4) or rhenium (5).
Specifically, reaction mechanisms and the fac- tors which influence the performance of vari- ous oxidants are poorly understood. It is clear, however, that several of the high valent 0x0
selective catalytic oxidant, which would not attack other potentially reactive groups, such as double bonds, halides, epoxides, esters, amides, or protecting groups, such as silyl or benzoate, amongst many others. Ideally, this catalytic oxi- dant would also operate at room temperature in an environmentally friendly solvent system (such as water) and use “green” co-oxidants such as oxygen ( O J or hydrogen peroxide (H202). This ideal catalytic oxidant should also be easy to recycle. Designing such systems remains a challenge, although significant progress is being made. Other factors which influence the acceptance of new catalysts include the ease of operation and the ease of (synthet- ically or commercially) producing the catalyst.
Ruthenium Oxide Catalyst
Systems
T h e only known well-defined stable ruthe- nium(VII1) compound is ruthenium tetroxide, RuO,, which is frequently far too vigorous an oxidant, as it attacks many functional groups, such as double bonds, and often forms car- boxylic acids. Consequently, research has focused on ruthenium compounds in slightly lower oxidation states.
“Catalytic” systems reported in the literature with reasonably high selectivities, conversions (depending on substrates) and turnovers (mea- sured as moles of product per mole of catalyst), are [Ru,0,(py),].3.5HI0 (py = pyridine), truns-[4-‘Bu-Hpy] [RuOlC1,(4-‘Bu-py)] (Hpy is a protonated pyridine), [LPh,] [RuO,Cl,] (L =
P, As) and truns-[Ru02(py),] [BF,]>, all having 4-methylmorpholine N-oxide, MNO, as co-oxi- dant (6-8). These compounds are obtained in moderate yield, and the first two are made by utilising RuO, as a gas, and require appropriate manipulative and experimental skills. The di- nuclear compound also appears to have a lim- ited “shelf-life”.
A series of compounds, which appear to have considerable potential as viable catalytic oxidants are the compounds cis-[PPh,]- [RuO,Cl,(OCOR)] (R = Me, Et, Pr, CHF,),
(9). The cis-0x0 arrangement is unusual because of the considerable electronic stabilisation asso- ciated with the trans-dioxo, d’, configuration
(10). The acetic acid derivative was shown to be a good catalytic oxidant with MNO as co-oxi- dant. No effects were observed on varying the length of the alkyl chain (R). It was felt that this system was worth further investigation and high yield routes to the compounds, where R = Me, CF,, C,,H,, C,F, and G H , , , were developed, which avoided the need to use RuO, gas. The compounds were found to be stable and cat- alytically active with a wide range of co-oxidants ( H 2 0 , , NaOCI, ‘BuOOH {tertiary-butyl hydroperoxide)) with MNO and tetrabutylam- monium periodate being the best, amongst oth- ers. Fluorinated compounds often display very different properties to their protonated ana- logues, probably because fluorine and hydro- gen, although being of similar size, have very different electronegativities. However, we found that apart from significantly affecting the syn- thetic yields of the oxidants, no effect on the oxidations was observed.
The most well-known ruthenium oxide cata- lyst, tetrapropylammonium perruthenate (TPAP) and its sister tetrabutylammonium per- ruthenate (TBAP) are effective in many selec- tive organic transformations of the type sum- marised in Equations (i) and (ii) using M N O as the co-oxidant. A comprehensive review has recently been published ( 1 1 ) . TPAP is also almost the only ruthenium oxidation catalyst to have been studied kinetically. The reaction of TPAP with propan-2-01 was found to be auto- catalytic, possibly due to the formation of col- loidal RuOz which may form RuO,mRuO, (1 2). TPAP is a three-electron oxidant overall, but it is believed that the alcohol oxidation step is of the two-electron type. The fact that TPAP will oxidise cyclobutanol to cyclobutanone sug- gests that a one-electron oxidation process is not involved, since a one-electron oxidant would cleave cyclobutanol to acyclic products ( 1 3 ) .
Other reported ruthenium(V1) oxidants will usually convert primary alcohols into acids, although internal alcohols will still be converted into ketones. Compounds reported to effect these transformations are K[RuO,] and [RuO,]’ (really tr~ns-[RuO,(OH)~]’ ) (7, 14, 15). T h e yields range from moderate to good, depending
0
Fig. 1 The proposed ester interme- diate in ruthcuiuni oxide catalysed
‘c=o
/
1 reactionsn
OH
on the substrate. Conversions are carried out in basic aqueous media, as ruthenate and per- ruthenate are unstable at lower pH. This lim- its the reagents to substrates which are stable at high pH. The reactions are rendered catalytic by using K&OR (persulfate) or BrO; (bromate) as the co-oxidant. Similar results are attained in the same media using tr~ns-[RuO,(HI0,)~]~ as catalyst, with NaIO, as the co-oxidant (16).
Even ruthenium tetroxide, RuO,, has been reported as oxidising secondary alcohols to ketones (1, 17). However, both RuO, and sodium ruthenate are known to attack double bonds, although sodium ruthenate will do so only if high temperatures or longer reaction times are employed ( 18, 19). Since RuO, is unstable at pH > 8, the active species in those reactions reported to involve RuO, and NaOCl is prob- ably RuO,-, see Equation (iv) (20).
4Ru0,
+
4 0 H+
4Ru0,+
2H,O+
O2 (iv)At high p H (> 12) though, ruthenate is the dominant species:
4 R u 0 ,
+
4 0 H+
4“RuO,”’+ 2 H 2 0+
0, (v)The compound [Ru02(tetramesitylporphyri- nato)] has been reported to oxidise alkyl alcohols to ketones, although its primary application is as an alkane oxidant (2 1).
Strictly speaking, few of the above mentioned ruthenium catalysts are true catalysts, since if the “catalytic” reactions are carried out with too little co-oxidant, the ruthenium compound remains in a reduced state. A catalyst should be unchanged after a reaction. Instead, the ruthe- nium 0x0 compounds are really primary oxi- dants, that is, they directly oxidise the substrate. The co-oxidant is the secondary oxidant - which
regenerates the primary oxidant. A few of the commonly used secondary oxidants do react directly with alcohols (for example, MNO with
primary alcohols), but the conversions are generally low.
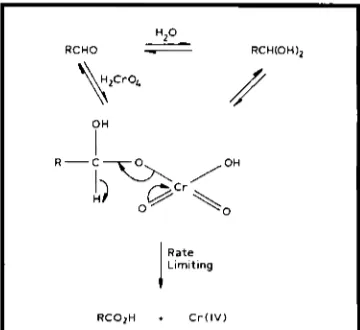
Alcohol oxidations with transition metal 0x0 reagents probably mainly involve the same basic transformation as shown in Figure 1, involv- ing an ester intermediate. For all of the catalytic oxidants mentioned above, primary alcohols react more rapidly than secondary alcohols, probably due to steric hindrance of secondary alcohols. It is generally thought that the reagents, reported above, which effectively catalyse the reaction in Equation (i), do so because of the presence of molecular sieves in the reaction medium. The molecular sieves efficiently remove water (formed in the reaction and present as water of crystallisation of MNO). It is known that an aldehyde hydrate is necessary for the reaction in Equation (iii) to occur. The mech- anism for the oxidation by chromic acid of an aldehyde is shown in Figure 2 (22) and ruthenium oxide oxidations are presumed to be similar. Some preliminary results from our
PH
R C O i H I C r ( l V )
OH
I
H-CRzOH
I
0 = Ru0;-
I
\
RzC=O + H z O
Fig. 3 Proposed mechanism for the oxidation of propan-2-01 by ruthenate and perruthenate
laboratory, however, imply that the presence of water in the reaction medium does not neces- sarily mean that the reaction in Equation (iii) will proceed.
The stoichiometric oxidations of propan-2-01 with the sodium salts of ruthenate and per- ruthenate have been investigated kinetically and mechanistically (23). Both reactions are believed to proceed via free radical-like transition states, although the subsequent steps differ. For both ruthenate and perruthenate, an ester interme- diate, as shown in Figure 1, is thought to be unlikely and instead it is assumed that the
ruthenium salts add to an
a-C-H
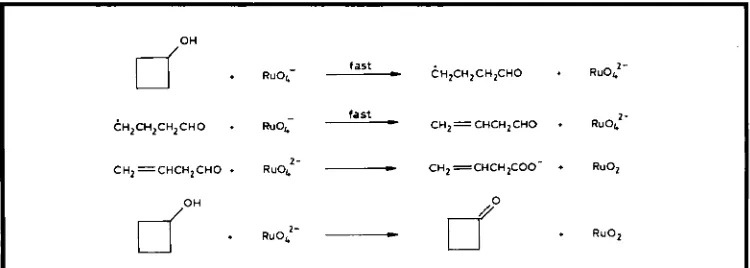
bond of the alcohol. The proposed mechanism is shown in Figure 3. Since HRu0,'- is very reactive, it is believed that a single electron transfer would occur very rapidly, before the radical could dif- fuse out of the solvent cage. For the stoichio- metric oxidation of propan-2-01 with per- ruthenate, as shown in Figure 3, it is thought likely that because the HRuO, ion is less reac- tive, the product would move out of the solvent cage before further reaction. These proposed mechanisms fit theoretical predictions (24).The reactions of [RuOJ and [Ru04]' with
fast
.
R u O c-
& i z ~RUO:- ~ z ~ ~ z ~ ~ ~fast
-
CHz=CHCHzCHO f RU0:-
doH
C H ~ C H ~ C H ~ C H O
.
R U O ~C H ~ = C H C H ~ C H O
.
R~O,, 2--
C H ~ = C H C H ~ ~ - + RuOZdoH
-
RuO:-do
+ RuOzFig. 4 Proposed mechanism for the one- and two-electron oxidation of eyclobutanol
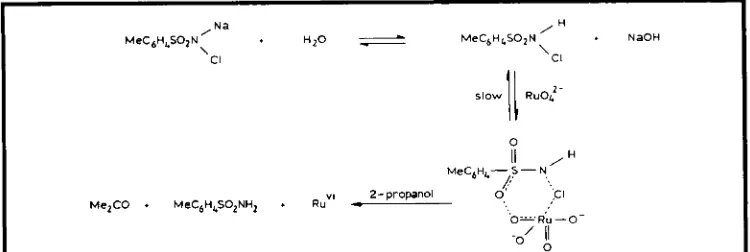
Na
MeC6H4S02N
’
.
H z O 5 MeCgHLSO2N / H + NaOHCI ‘Cl
slow
11
RuOJMelCO MeC6H4SOZNH2 R””’
0 II ,H
MeCgHL- 5 - N,
/
’.,* 2- propanol o., , .c I
Fig. 5 Proposed mechanism for the activation of rutlienate 11y chloraniine-T, prior to the oxidation of propan-2-01
I
cyclobutanol suggest that the latter is a nvo-elec- tron and the former, unlike TPAP, is a one- electron oxidant, see Figure 4 (25).
The mechanisms of the catalytic oxidations of propan-2-01 using [RuO,]’ with a variety of co-oxidants have also been investigated. For the co-oxidants hexacyanoferrate(III), diperioda- tocuprate(II1) and periodate, the data imply that a complex is formed, as for the stoichiometric oxidation, Figure 3 . The role of the co-oxidant is only to regenerate Ru(V1). The exception is when the co-oxidant is chloramine-T. In this case, the co-oxidant appears to react directly with the Ru(V1) ion, see Figure 5 . This active complex then reacts rapidly with propan-2-01 to form acetone (26). It seems very likely, how- ever, that these mechanisms are only valid in aqueous base and the mechanisms are proba- bly different in non-aqueous media, as shown, for example, by the different reaction prod- ucts obtained in the oxidation of cyclobutanol with TPAP and basic perruthenate.
The disadvantages of many of the catalysts that catalyse the reaction in Equation (i) and also of several which efficiently catalyse the reac- tion in Equation (ii) are that they require CHZC12 or C H C N as solvents (which are not “green”) and that they require M N O as co-oxidant. However, the MNO co-oxidant is expensive, an irritant to the eyes, the respiratory system and the skin, and its final product, 4-methyl mor- pholine, is corrosive and causes burns to the skin, the eyes and mucous layers in the nasal
tract. Apart from this, even the best known catalysts do not give quantitative or almost-quan- titative transformations in all apparently suit- able systems and, as such, there is a great need to develop more effective and more selective catalysts with higher turnovers.
In general, the activity of catalysts is affected by the steric bulk (for instance, cone angles) of the attached groups and the electron density on the metal. The significant effect of the groups attached to the Ru(V1) centre is demonstrated by the examples of Ru(VI)O, moieties bonded to porphyrins or to other bulky tetradentate lig- ands: many of these compounds are effective catalysts for the epoxidation of olefins (27). None of the other ruthenium(V1) 0x0 catalysts, referred to above, can perform this reaction. However, even Ru(VI)02 compounds, with bulky bidentate ligands, can catalyse epoxida- tion reactions (28).
Towards Commercial Catalysts
(a) (b)
a r b - Br6-
"c606- '6 ; T B r 6 -
6- -"6</) / 6 . 1 6 -
6. Ru 6. 6'
* C F
Ob- 6.
(C 1 ( d )
Fig. 6 Loealised relative charges on (a) 3-Br-pyridine and (b) 4-Br-pyridine and the resulting relative charge distribution in the complexes they constitute (c) and (d), respectively
mono-oxo ruthenium(V1) organometallic com- pounds are known (29) and ruthenium(V1) organometallic di-oxo compounds have so far proved to be totally elusive.
Alternatively, one can study the effects of sub- stituents in inorganic ruthenium 0x0 com- pounds. This latter approach has the advantage that routes to discreet, fully characterisable com- pounds of this type are already available.
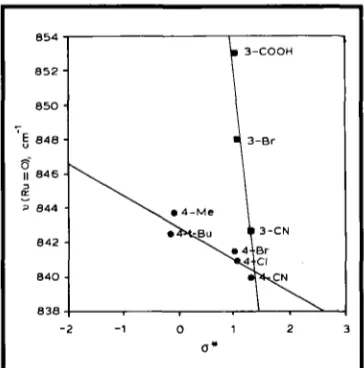
The first system to be considered consisted of compounds of the type [RuO,Clz(Y-py),] (Y = H, 4-'Bu, 3-CN, 4-CN, 2-Br, 3-Br, 4-Br, 4-C1, 4-Me, 4-C(0)C,H5, 3-COOH, 4-COOH). This series was chosen because we thought that sub- stituents on a flat aromatic ring, for which Hammett substituent constants were available, should show easily quantifiable electronic effects on the ruthenium centre. The first examples of compounds in this series Cy = H, 4-'Bu, 4-C1)
were prepared by Griffith and co-workers (6) and the rest by us (30). As can be seen from Figure 6, substitution at either the 3- or 4-posi- tion of the pyridine ring should have a signifi- cant effect on the charge on the pyridine nitro- gen and consequently on the net charge on the ruthenium centre.
Similarly, the nature of the substituent Y (either electron pushing or electron withdrawing) should affect the charge on the pyridine nitrogen and
hence on the ruthenium centre. These effects should be revealed by changes in the Ru=O
stretching vibrations. This is confirmed by exam- ining the IR spectra of these compounds.
3-COOH
850
6
8 4 86
II 8 4 6 -
z
-
3 844
-
842 -
8 4 0 -
8 3 8 -
- 2 - 1 0 1 2 3
o*
Fig. 7 A plot of v(Ku=O) values against O* for the compounds [ R u O , C I , ( Y - ~ ~ ) ~ ] ; O* is a description of the substituents. The magnitude of O* gives the relative strength of the electron- withdrawing or electrotdonating properties of the substituents. The value of O* is positive if the substituent is electron-withdrawing and negative if it i s electron-donating
Stoichiometric and *Catalytic Oxiclatioiis of I-Hexaiiol to Hexaiial by (Y)O,CI,Ru with a Range of Co-catalysts, over a 24-hour period
Co-catalvsts. O/O conversion
Substituted
pyridine (Y)
Characteristic ;toichiometric
oxidations
“NaOCI “‘BuOOH
39 34 38 84 63 59 50 50 63 7 - “MNO 31 *H20, 15 8 10 15 45 36 - - 13 6 ~ - ~ -
v(Ru-0). c m
13 9 11 47 85 64 65 45 17 6 ~ - ~ ~ - 12 9 10 39 55 51 - - ~ 15 5 - ~ ~ ~ 22 14 16 54 97 99 96 65 23 16 ~ - ~ ~ ~ 842.4(vs) 842.7( m ) 840(vs); 831(s)
826(s) 848(vs); 817.7(s)
841 . I (vs) 840.5(vs) 849.3 m ) 837.5(vs) 839(vs) 845.5ivs) 843.7(vs); 830.9(s)
840.8(s) 853( s )
841.5(m)
All runs wero icpeated at ledst three ;lines and are reprodiicihie ni
Excluding the compound where Y = H, the v(Ru=O) stretches correlate linearly with the Hammett, o,, and Taft, o*, values, see Figure
7, and Pauling type electronegativity (X) values of the Y substituents. Since these values are due to an inductive effect, one would expect the inductive effect to increase with decreasing dis- tance between the substituent, Y, and the ruthe- nium centre. This is observed, with the slope of the graph for the 3-substituted compounds being much steeper than for the 4-substituted compounds.
Thus, these complexes are stoichiometric and “catalytic” oxidants for the conversion of alco- hols to aldehydes, with the substituents, Y, clearly affecting the performance of the compounds. The Table shows values for stoichiometric oxi- dations of 1-hexanol to hexanal by (Y)0,Cl2Ru with a range of co-catalysts.
A sequence was observed of the oxidising abil- ity for the stoichiometric oxidations: phen < 4-Me-py < benzoyl-py < 3-CN-py < 4-CN-py < 4-IBu-p~ < 4-C1-pyz py
-=
3-Br-py= 4-Br-py < bipy. An almost identical sequence was~ niediurn. s ~ strong, vs = vcry strong
observed for the “catalytic” oxidations, using H,O,, hypochlorite (OC1) or t-butyl hydroper- oxide ((CH,) C O O H ) as the co-oxidants. Although the Y substituents clearly affect the v(Ru=O) values and hence the strength of the Ru=O bond and the electron density on the metal, no correlation between oxidising ability and substituent constants for Y is observed. This implies that electron density on the metal has no significant effect on these compounds as oxidants.
In addition, electronic effects do not appear to play a role in the performance of the Ru(V1) salts, [PPh,] [Ru02C12(0COR)], as reported above. While most of the above compounds are mediocre oxidants, the performance of
[RuOZC1,(4-Br-py),] is exceptional and so is that of [RuOzC12(3-Br-py)J, to a lesser extent. Both of these are far superior oxidants to any of the other compounds in this series. The 4-Br-pyri- dine derivative gives especially high rates of con- version with reasonable turnovers. We are at present investigating this clear ligand effect.
bipyridyl, 1,lO-phenanthroline) indicates that the rigidity of the ligands may also play a role, as the bipyridyl compound is a reasonable oxi- dant, whereas the phenanthroline compound is a very poor oxidant, as shown in the Table.
One of the great disadvantages of many homo- geneous catalytic systems is the problem of sep- arating the product from the catalyst. For this reason heterogeneous catalysts remain very pop- ular, as the catalyst can simply be filtered off and reused.
One way of attempting to combine the advan- tages of homogeneous and heterogeneous cat- alysts is by bonding soluble catalysts to insolu- ble supports, although these have yet to attain a large scale industrial breakthrough (31). We have supported ruthenate on poly-(4-vinylpyri- dine) and on zeolite-Y. Both these supported catalysts appear to be very selective in the oxi- dations referred to in Equations (i) and (ii), with a wide range of co-oxidants at room tempera- ture (32). The catalysts can be filtered off and reused. We are currently investigating both the structures and the versatility of these catalysts. We believe that the ruthenate may be bonded to the polymer, as is the OsO4/poly-(4-vinylppi- dine) system for oxidising alkenes to diols, rather than just being supported ( 3 3 ) .
A ruthenium hydrotalcite heterogeneous com- petitor has recently been reported (34). This catalyst is reported to oxidise primary and sec- ondary alcohols into aldehydes and ketones, respectively, in high yields with high selectivity. This catalyst appears to be easily prepared and the oxidations take place at 60°C in toluene, using molecular oxygen as co-oxidant. It would appear that a heteronuclear interaction between ruthenium and cobalt is responsible for the high activity, with ruthenium probably being oxidised
to a + 6 oxidation state within the Brucite, Mg(OH)?, layer.
The use of oxygen as co-oxidant, as in the sys- tem mentioned above, is very advantageous kom an environmental point of view. We have car- ried out extensive studies on various co-oxidants for the catalysts [ R U O ~ C ~ ~ ( Y - ~ ~ ) ~ ] , [PPh,]- [RuO,Cl,(OCOR)] and polymer or zeolite “sup- ported” ruthenate. While these systems are active for a wide range of co-oxidants, consistently, highest conversions for these systems and many of those reported above, are usually obtained using MNO, although tetrabutyl-ammonium periodate appears to be an excellent co-oxidant for the immobilised systems and also for the oxi- dation of primary alcohols with the salts [PPh,] [RuO?Cl2(0COR)] (R = CF1, C,H, and C,F,).
Concluding Remarks
Thus, it would seem that high oxidation state ruthenium catalysts are amongst the best mild oxidants for the oxidation of alcohols, with little or no over-oxidation products being obtained. Our information remains limited on the mech- anism of the reactions and on which factors, besides having ruthenium in a high oxidation state, influence the efficiency of these compounds. Our best hope of discovering what influences the oxidation chemistry of ruthenium will come kom using homogeneous catalytic systems, although environmental and economic concerns may ultimately favour supported, heterogeneous or genuinely two-phase catalytic systems.
Acknowledgement
Thanks to the Alexander von Humboldt Foundation, N M T Electrodes, Western Platinum Refineries, the University of Natal and the Foundation for Research Development for their support; also to Vikash Gokul.
References 1 R. A. Sheldon and J. K. Kochi, “Metal Catalysed
Oxidations of Organic Comoounds”. Academic
5 A. Gansauer, Angew. Chem. Inr. Ed., 1997, 36,
259 1 and references therein Press, New York,l981
B. C. L. Weedon,J. Chem. SOC., 1946, 39
2 K, Bowden, 1. M. Heilbron, E, R. H. Jones and A. C. A. O’Mahoney and D. J. Williams,
c.
A. M. E1-Hendawy, w.3.
Grifith, Chem.SOC., Dalton Trans., 1990, 737
3 A. J. Mancuso and D. Swern, Synthesis, 1981, 165 4 T. Gobel and K. B. Sharpless, Angew. Chem. Int. Ed., 1997, 36, 2591 and references therein; M. Schroder, Chem. Rev., 1980, 80, 187
7 A. M. El-Hendawy, W. P. Grifith, F. I. Taha and M. N. Moussa,J Chem. SOC., Dalton Trans., 1989, 90 1
8 G. Green, W. P. Griffith, D. M. Hollinshead, S . V. Ley and M. Schroedcr, J. Cliein. Sor., I’erkirr E m s . I , 1984, 681; A. C. Dengel, W. P. Griffith, A. M. El-Hendawy and J. M. Jolliffe, l’iily/~edri~ii,
1990, 9, 1751
9 W. P. Griffith, J. M. Jolliffe, S. V. Ley and I). J.
Williams,
.7.
Ckenr. Soc., Uteri/. Co~iiir/uir., 1990,12 19; W. P. Griffith and J. M. Jolliffe, J. Cheni.
SUC., Uulroir Trans., 1992, 3483
10 A. Dovletoglou, S. A. Adeyenii, M. H. Lynn, D. J. Hodgson and T. J. Meyer, J . Ani. Chrwr. ISoc.,
1990, 112, 8989 and references therein
I 1 S. V. Ley, J. Norman, W. P. Griffith and S . 1’.
Marsden, S ~ ~ i i r h e s i s , 1994, 639
12 D. G. Lee, Z . Wang and W. D. Chandler,.y. Org. Chein., 1992, 51, 3276
13 J. Rocek and C . 3 . Ng, J. Am. C h m . Sor., 1974, 96, 1522
14 A. J. Bailey, W. P. Grifith, S. I. Mostafa and P. A. Sherwood, Iiiurg, Clieni., 1993, 32, 268 15 D. G. Lee, D. T. Hall and J. H. Clcland, C t r i i .
-7.
Chenr., 1972, 50, 3741
16 A. M. El-Hendawy, W. P. Griffth, B. Piggott and D. J. Williams,.7. Chcirr. Soc., Dalroii h i i s . , 1988, 1983
17 P. E. Morris, Jr. and D. E. Kiely, J. O r g C X e n i . ,
1987, 52, 1149; S. Tori, T. Inokuchi and T.
Sugiura,J. Org. (,‘liein., 1986, 51, 155
18 P. H. J. Carlsen, T. Katsuki, V. S. Martin and I<. B. Sharplcss,J. O r g Clzenr., 1981, 46, 3936 19 D. G. Lee, S. Helliwell and V. S. Chang,J. Chg
Chenl., 1976, 41, 3646
20 A. Mills and C . Holland,.7. O’ireiii. Res., 1997, 36s;
R. E. Connick and R. Hurlry,,7. Atti. C X e i n . Soc.,
1952, 74, 5012
2 1 H. Ohtake, T. Higuchi and M. Hirobe, J. Ani. Chztrl. Soc., 1992, 114, 10660
22 J. RocekandC.-S. Ng,J Or$. Chern., 1973,38,3348 23 D. G. I x e and I . . N. Congson, C h i . J. Cheni.,
1990,68, 1774
24 A. K. Rappt and W. A. Goddard III,J. A W L Chenl. SOL-., 1982, 104,448; 3287; K. Behari, N. Narayan, R. S. Shukla and I(. C. Gupta, h i .
.7.
Ch~.ni. Kinet.,1984,16, 195
25 D. G. I.ee, I.. N. Congson, U. A. Spitzer and M. E. Olson, C u i i . J . Cheni., 1984, 62, 1835 26 T. V. L,alitha and B. Sethuram, Transirion Mcr.
Chori., 1992, 17, 29
27 Z. Gross and S. Ini,J. Org. C,’heni., 1997,62, 5514;
C.-M. Che, C.-K. I.i, W.-T. Tang and W.-Y. Yu,
J. C h i / . SOC., DU/Z<JY/ Trails., 1992, 3153; J. T.
Groves and R. Quinn, J. ,4nl. C h t Suc., ~ 1985, 107, 5790
28 C, I-, Bailey and R. S . Drago,J. C h i . SLY., Chcrri.
C o i i i t i z i o / . , 1987, 179
29 R. W. Marshman, W. S. Bigham, S . R. Wilson and P. A. Shapley, Org~r~rr~nrrtuNi~~s, 1990, 9, 1341 30 H. 13. Friedrich, E. Friedrich, V. Gokul, K. K.
Kubhcka and N. Rinder, submitted to S. A.fr. j?
Cheni., in press
3 1 W. A. Herrniann, Kuriiukre (Uuritlsiud~, 1991,29 32 N. Singh and H. B. Friedrich, Abstr. 1 Ith
International Symposium on Homogeneous Catalysis, St Andrews, Scotland, July, 1998, 261 33 W. A. Herrniann, R. M. Kratzer, J. Bliimel, H. B. Friedrich, R. W. Fischer, D. C. Apprrley, J. Mink and 0. Bcrkesi, J. Mol. Curd., 1997, 120, 197 34 T. Matsushita, K. Ebitabi and K. Kaneda,J. Clienr.
S O C . , < ; / i d / i i , C h r r r i / / i i / . , 1999, 265
Ruthenium
in
Living Radical Polymerisation
Living radical polymerisation has become a n presence and absence of aluminium(II1) important technique in precision polymer syn-
thesis for the construction of new polymers with tailor-made structural complexities. In order to assemble structurally well-defined polymers, in terms of molecular weights and molecular weight
isopropoxide, Al(0z-Pr) ,, at 80°C (T. Nishikawa, M. Kaniigaito and M. Sawamoto, Mucronzolecules,
1999, 32, (7), 2204-2209).
With ruthenium(I1)-initiation, living radical suspension polymerisation of MMA proceeded distributions, their polymerisation must be con-
trolled. However, the control of the reactivity
of the propagating chain ends was difficult, until the recent discovery of living (controlled) rad- ical polymerisation. D u e to its unique tolerance t o water, radical polymerisation c a n be per- formed as suspension, dispersion and emulsion processes.
Most of these living radical polymerisations are based o n transition metal catalysis (via reversible activation of a carbon-halogen ter-
to give poly(MMA) with controlled high mol- ecular weights @,,) and narrow molecular weight distributions
(M>s
/M,,
= 1 . 1 to 1.3). In water, the PhCOCHCl,iKuCl1(PPhi), initiating sys- tem gave poly(MMA) of M,,-
10’ andM\\
lmn-
I . 1, even in the absence of Al(0i-Pr) $.
Polymerisations were faster in water than in organic solvents, such as toluene. T h e radical mechanism of the polymerisation was confirmed and the high stabilities of the dormant carbon- halogen terminal and of the ruthenium catalyst minal) or nitroxide-mediated processes.
Recently, workers at Kyoto University, Japan, have examined t h e living radical suspension polymerisation of methyl methacrylate (MMA)
in water and alcohols using RuCI,(PPh,) catalystiactivator and a n organic halide initia- tor, such as P h C O C H C l , o r CCI,Br, in t h e
i n water and alcohols were demonstrated. Ruthenium complexes are known to be weakly oxophilic and sometimes water tolerant.