Genetic Labelling and
Transplantation of Schwann cells to
Enhance Peripheral Nerve
Regeneration
Mr. AFSHIN MOSAHEBI-MOHAMMADI
MBBS FRCS
Doctor of Philosophy (Plastic Surgery)
Royal Free & University College Medical School
ProQuest Number: U642240
All rights reserved
INFORMATION TO ALL USERS
The quality of this reproduction is dependent upon the quality of the copy submitted.
In the unlikely event that the author did not send a complete manuscript and there are missing pages, these will be noted. Also, if material had to be removed,
a note will indicate the deletion.
uest.
ProQuest U642240
Published by ProQuest LLC(2015). Copyright of the Dissertation is held by the Author.
All rights reserved.
This work is protected against unauthorized copying under Title 17, United States Code. Microform Edition © ProQuest LLC.
ProQuest LLC
789 East Eisenhower Parkway P.O. Box 1346
“The worst fool is certain o f his
i
I
I
A n cie n t Persian proverb
^
Knowieage
g
I
I
I
I
^
“i am certain of nothing but the holiness o f
i
I
I
i
the heart’s affections, and the truth
of
i
Abstract of thesis
There is a clinical need to improve functional outcome after peripheral nerve
reconstruction, and also to find a replacement for autologous nerve grafts
currently used for this purpose. Schwann cells (SC) are essential for adequate
axonal regeneration and the aim of this work was to develop a methodology to
enable investigation and characterisation of the effects of SC transplantation
and genetic manipulation on peripheral nerve regeneration.
Initially an identifiable and pure population of cultured SC was obtained. In
vitro evaluation showed that chemical labelling adversely affected SC
properties. Transduction of lacZ genetic label was carried out and a stable
population of genetically modified SC was obtained. Transduced SC
properties and lacZ expression were preserved in vitro for 6 months of
continuous culture. Suspension matrix is required for SC transplantation and
the suitability of alginate hydrogel was confirmed by in vitro tests to support
SC proliferation and neurite sprouting in a neuron-glial co-culture.
Defects in the rat sciatic nerve were bridged using resorbable poly-3-
hydroxybutyrate (PHB) conduits containing SC. The results showed that the
optimal number of SC required to enhance axonal regeneration was
80x1 oVml. Following transplantation of transduced SC, syngeneic SC could
be identified for up to 6 weeks and allogeneic SC for up to 3 weeks, as
identified by X-gal staining. Immunohistochemistiy was used to characterise
SC, immune response, and axonal regeneration and the staining quantified by
image analysis. Transplantation of both syngeneic and allogeneic SC
improved axonal regeneration distance, the quantity of regeneration was better
and more sustained with syngeneic SC. Furthermore, addition of liquid
fibronectin to alginate improved regeneration which was further enhanced
when SC were present. Finally a new technique for nonviral gene delivery of
insulin-like growth factor I and mechano growth factor gene with alginate
hydrogel matrix showed promising results in improving peripheral nerve
regeneration.
Acknowledgement
I would like to thank my parents for instilling the seeds of curiosity in me and
for their continual love, support and encouragement in every step of my life. I
would also like to thank my sister for her patience, support and understanding.
Similarly, I thank my partner Mitra for her encouragement in the last stages of
this work.
This work would have been impossible without the dedication and guidance
of my supervisor, teacher and friend Dr. Giorgio Terenghi. I would like to
express my deepest gratitude to him for teaching me to look at scientific basis
behind the clinical problems. Dr. Barbara Woodward overlooked the initial
stages of my work with Schwann cells and her enormous expertise and
assistance was invaluable. I would also like to thank the past Director Prof.
Colin Green and also Dr. Robin Martin for giving me the opportunity to work
at Blond Mclndoe and for their advice and encouragement. All other staff at
the Blond Mclndoe centre also helped in different ways during this project.
Dawn Field, Keith Wood, Hilary Morely, Lesley Taylor and our fundraiser
Caroline Leet.
The smooth transition to Royal Free campus was possible through the efforts
of Prof. Marc Winslet and Mrs. Karen Cheetham. Richard Young, Andrew
Hart and Stuart James, Magda Simon and Pari Mohana created a fun
environment in the laboratory. Paul Fuller kept the lab organised and was
helpful in the analysis of my work. My thanks to Dr. Sas Dijk, Department of
Anatomy, for his help in preparation of plasmid cDNA and Prof. G Goldspink
for allowing me to use the gene for MGF. I would like to thank Mr. Peter
Butler and Prof. Gus MoGrouther for their advice and encouragement
throughout. I thank Mr. Simon Kay and Mr. Harry Belchor for their assistance
in the collection of human nerve samples.
I am gratefiil to the Smith's Charity and the Geoffrey Burton Charitable Trust
for their generous financial support. I would like to thank Dr. David Gwynne,
Cambridge NeuroScience, Cambridge MA, USA, for the gift of recombinant
human glial growth factor, Astra Tech for PHB and Pronova for alginate.
Last but not least my special gratitude goes to Prof. Mikael Wiberg for his
extraordinary energy, efforts and guidance in this work
Table of contents
Abstract of thesis
3Acknowledgements
5Table of Figures
14Chapter 1: Introduction
L l Morphology o f the peripheral nerve 18
1.1.1 Neurons 18
1.1.2 Schwann cells 21 1.1.3 Extracellular matrix 24
L2 Axonal degeneration 25
1.2.1 Classification o f nerve injury 25 1.2.2 Wallerian degeneration 26
L3 Axonal regeneration 28
1.3.1 Role o f Schwann cells 29
L4 Peripheral nerve repair 30
1.4.1 Nerve gap reconstruction 32 1.4.1.1 Nerve graft 32 1.4.1.2 Conduits and other alternative methods 33 1.4.2 Time factor 36
L5 Bioengineered nerve conduits 37
1.5.1 Transplantation o f cultured Schwann cells 38 1.5.1.1 Schwann cell chemical labelling 40 1.5.1.2 Schwann cell genetic labelling 41
1.5.2 Matrix 42
1.5.2.1 Alginate 43
1.5.3 Role of gene therapy in nerve repair 46
1.6 Hypothesis 49
1.7 Aims 49
Chapter 2; Materials and Methods
2.1 Rat Schwann cell culture 52
2.1.1 Nerve harvesting 5 2 2.1.2 Digestion 52 2.1.3 Purification & maintenance 54
2.2 Human Schwann cell culture 55
2.2.1 Nerve collection & digestion 55 2.2.2 Purification & maintenance 57
2.3 Other tissue culture methods 58
2.3.1 Rat perineural fibroblast culture 5 8 2.3.2 Chick embryo dorsal root ganglion culture 59 2.3.3 PT67 packaging cell line culture and maintenance 61 2.3.4 Cryopreservation & defrosting o f cells 61
2.4 Cell viability assays 62
2.4.1 MTT assay 62 2.4.2 Alamar bluerM assay 62
2.5 Chemical cell labelling 63
2.5.1 H33342 63
2.5.2 PKH26 63
2.5.3 Co-culture o f double labelled Schwann cells 64
2.6 Genetic manipulation 65
2.6.1 PT67 retroviral packaging cell line selection 65
2.6.2 Retroviral transduction by supernatant transfer 66 2.6.3 Screening for retroviral production from transduced Schwann cells 67 2.6.4 Transduced cell detection 69 2.6.4.1 Chemical staining 69 2.6.4.2 Preparation of alginate stock 70 2.6.5 Plasmid DNA delivery 71
2.7 Matrices for Schwann cell suspension 71
2.7.1 Alginate gel 71 2.7.1.1 Preparation of alginate stock 71 2.7.1.2 Dilution of stock and gel setting 72 2.7.1.3 Cell suspension 72 2.7.1.4 Cell recovery from alginate 73 2.7.1.5 Cryopreservation of alginate/ cell mixture 73 2.7.1.6 Visualisation of alginate gel 73 2.7.1.7 Alginate strands preparation 74
2.7.2 Collagen gel 74
2.7.3 Fibrin glue 75 2.7.4 Hyaluronic acid 75 2.7.5 Matrigel 75
2.8 Poly hydroxy butyrate (PHB) 76
2.8.1 PHB conduits 76
2.8.2 Schwann cell culture with PHB fibres 78
2.8.3 Loading o f PHB conduit with alginate mixture 78 2.8.4 Coating o f PHB conduit with alginate 78
2.9 Operative procedure 80
2.9.1 Anaesthesia, pre-operative preparation, recovery & euthanasia 80 2.9.2 Wrap around repair 80 2.9.3 Gap repair 82 2.9.4 Nerve and conduit collection & processing 82
2.10 Morphological assessment 84
2.10.1 Immimostaining of cultured SC 84
2.10.1.1 SlOO immunostaining 85 2.10.1.2 Staining for GFAP/ LNGFR, p75/ MHC1/ MHCII 85
2.10.2 Immunostaining of sections 86
2.10.2.1 SlOO/PanFJF immunostaining 86 2.10.2.2 Staining for NCAM/ MBP/ CD2/ CD45R/ ED 1/ p75/ MHC I & H 86 2.10.2.3 Combined X-gal and immunostaining 87
2.10.3 Immunostaining of cultured chick DRG 87
2.11 Quantification 87
2.11.1 Tissue culture samples 88
2.11.1.1 Schwann cell transduction 88 2.11.1.2 DRG neurite growth 89
2.11.2 Axonal regeneration 89
2.11.2.1 Regeneration distance 89 2.11.2.2 Regeneration quantity: 90
2.11.3 Schwann cell quantification 91
2.11.3.1 Schwann cell ingrowth into the conduit 91 2.11.3.2 Schwann cell phenotype expression 91
2.11.4 Immunological assessment 92
2.11.4.1 B and T lymphocytes and macrophages 92 2.11.4.2 MHC class I and II expression 92
2.11.5 Statistical analysis 93
Chapter 3: Chemical labelling of Schwann cells
3.1 Introduction 95
3.2 Materials and Methods 96
3.3 Results 97
3.4 Discussion 106
Chapter 4; Genetic manipulation of Schwann cells
4.1 Introduction 111
4.2 Materials & Methods 111
4.3 Results 112
4.4 Discussion 120
Chapter 5: Schwann cell compatibility studies
5.1 Introduction 124
5.2 Materials & Methods 124
5.3 Results & analyses 125
5.3.1 Schwann cell and PHB fibres 125 5.3.2 Effect of calcium concentration on Schwann cell 127 5.3.3 Conqjarison of matrices 127 5.3.4 Neuronal culture in alginate 133
5.4 Discussion 135
Chapter 6: Variables of composite conduits
6,1 Introduction 141
6,2 Materials and Methods 142
6.2.1 Variable Schwann cell concentration 142 6.2.2 Alginate strands 142 6.2.3 Perineurial fibroblasts 142
6,3 Results 144
6.3.1 Optimal concentration of Schwann cells 144 6.3.2 Effect of alginate strands 149 6.3.2 Effect of perineurial fibroblasts 150
6,4 Discussion 151
Chapter 7: Syngeneic Schwann cell transplantation and
modulation of their matrix
7.1 Introduction 156
7.2 Materials and methods 157
7.3 Results 157
7.4 Discussion 165
Chapter 8: Allogeneic Schwann cell transplantation
8.1 Introduction 169
8.2 Materials and methods 169
8.3 Results 170
8.3.1 Transplanted Schwann cell 170 8.3.2 Regeneration 172 8.3.3 Schwann cell phenotype 175 8.3.4 MHC quantification 178 8.3.5 Immune reaction infiltrate 179
8.4 Discussion 183
Chapter 9: Gene delivery for peripheral nerve
regeneration
9.1 Introduction 189
9.2 Materials and methods 190
9.3 Results 190
9.4 Discussion 196
Chapter 10: Conclusions and future work
200
Publications
205
References
208
Appendices
241
Table of Figures
Chapter 1 :
1.1 Portrait of T. Schwann 21
1.2 Original drawings of T. Schwann 21
1.3 Extraction process of alginate 44
1.4 Molecular structure of alginate 45
Chapter 2:
2.1 Harvesting of rat sciatic nerve 53
2.2A Rat SC in culture 56
2.2B Human SC in culture 58
2.3 Perineurial fibroblasts 59
2.4 Exposed chick embryo spinal column 60
2.5 Transduction process 68
2.6 Preparation of alginate strands 74
2.7 Separating PHB sheets 77
2.8 Rolling & welding PHB conduit 77
2.9 S.E.M. of PHB sheets after welding 77
2.10A &B Filling of PHB conduit 79
2.11 Coating of PHB 79
2.12 Sciatic nerve injury model 81
2.13 PHB conduit secured in place 83
2.14 Harvested PHB conduit 83
2.15 Image capturing protocol for quantification 91
Chapter 3:
3.1 Double chemical labelling of SC 98
3.2 Rat SC co-culture (1 week) 99
3.3 Rat SC co-culture (4 weeks) 100
3.4 Differentially labelled co-culture ( 1 week) 102
3.5 Differentially labelled co-culture (4 weeks) 103
3.6A MTT ratio of chemically labelled SC 105
3.6B MTT ratio of chemically labelled mixed SC 106
3.7A&B GFAP staining at 1 and 2 weeks 107
Chapter 4:
4.1 Immunostaining of transduced rat SC 113
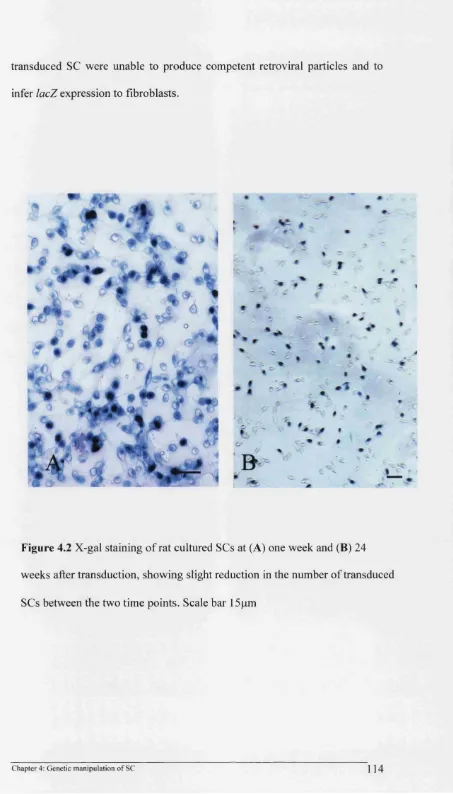
4.2 X-gal staining of rat SC 114
4.3 Non-transduced rat SC 115
4.4 Bar chart of % X-gal staining of rat SC 116
4.5 MTT ratio of transduced rat SC 117
4.6 Different level of marker expression 118
Chapter 5:
5.1 Alignment of SC along PHB fibre 126
5.2 MTT ratio of SC in different calcium concentrations 128
5.3 AlamarBlue assay of SC in different matrices 129
5.4 SC in alginate hydrogel 131
5.5 ESEM of alginate with PHB and SC 132
5.6 Neurite growth fi*om DRG culture in alginate 133
5.7 Bar chart of mean axonal sprouting from DRG 134
Chapter 6:
6.1 Implantation of alginate strands 143
6.2 X-gal staining of section through PHB conduit 144
6.3 Combined X-gal & immunostaining of section 146
6.4 Bar chart of axonal regeneration distance 147
6.5 Bar chart of axonal regeneration quantity 148
6.6 Effect of alginate strand 149
6.7 Transplanted fibroblasts 150
6.8 Effect of perineurial fibroblasts 151
Chapter 7:
7.1 AlamarBlue assay of SC in alginate
7.2 Different level of X-gal staining
7.3 Combined X-gal and immunostaining
158
160
161
7.4 Regeneration distance depending in different PHB conduits 163
7.5 Regeneration quantity depending in different PHB conduits 164
Chapter 8:
8.1 Different level of X-gal staining 171
8.2 Regeneration distance in conduits 172
8.3 Regeneration quantity in conduits 174
8.4 Quantity of SC ingrowth in conduits 174
8.5 Quantification of p75 staining 175
8.6 Quantification of NCAM staining 176
8.7 NCAM & p75 immunostaining 177
8.8 Quantification of MHC I & II staining 178
8.9 T-cell count in conduits 180
8.10 B-cell count in conduits 180
8.11 immunostaining for B & T-cells and macrophages 181
8.12 Macrophage cell count in conduits 182
Chapter 9:
9.1 Analysis of release of plasmids firom alginate 191
9.2 S100 staining of conduits 193
9.3 PanNF staining of conduits 194
9.4 Combined SlOO and PanNF staining of conduits 195
c
hapter One
1.1
Morphology of the peripheral nerve
Historical knowledge of the peripheral nerve dates back to 1000 B.C. as it has
been suggested that the description and the functions of the vagus and the
recurrent laryngeal nerves had been known in Sushruta. Hippocrates has been
credited with the first written description of the nerve and Galen was the first
to describe the effects of nerve transection. However, our present
understanding of the structure of the peripheral nerve stems from pioneering
works by van Leeuwenhoek, Fontana, Bell, Purkinje, Waller and Schwann
(Terzis & Smith 1990).
The peripheral nervous system is comprised of the peripheral nerve fibres and
their associated tissues, which include glial cells, sensory organs, muscle
motor end plates, intraneural vasculature and related extracellular matrix
(Landon 1976; Dyck & Thomas 1984). The focus of this chapter is on
neurons, glial cells and their extracellular matrix.
1.1.1 Neurons
The neuronal cell body is the site of all metabohc activities, which are then
transported along the axon. Motor neurons innervate striated muscles, and
their cell bodies and dendrites are located in the anterior horn of the spinal
cord and are often referred to as anterior horn cells. The axons pass through
the anterior root to join the peripheral nerve bundle. These axons are
myelinated except at their origins and terminations, where they divide into
numerous branches to form neuromuscular synapses. Motor neurons
innervating smooth muscles and glands belong to the autonomic nervous
system. Their cell bodies are found in the peripheral autonomic ganglia and
their axons are unmyelinated. Sensory neurons are located outside, but close,
to the spinal cord in dorsal root ganglia of the spinal nerves. These are
unipolar neurons and carry impulses generated from the skin as well as muscle
spindles, viscera and blood vessels back to the spinal cord. The sensory fibres
may be myelinated or unmyelinated according to the sensory modality.
A nerve fibre is generally described as any process of the nerve cell, either
dendrite or axon. It is a cylindrical extension of the cell soma, which arises
from the funnel shaped axonal hillock. A specialised membrane, axolemma,
surrounds it and maintains a resting potential by selectively altering the
concentration of monovalent ions. The nerve fibres can be divided into motor,
sensory and sympathetic, and are classified according to their fibre diameter
and conduction velocity into A, B and C fibres (Gasser & Grundfest 1939).
The principle content of the fibres, axoplasm, are microfilaments,
neurofilaments and neiu'otubules. Microfilaments, as their name implies, are
the smallest component and are made up of actin proteins and may play a role
in intra-axonal transport (Varon & Bunge 1978). Neurofilaments are larger
and more prevalent and are composed of proteins of 3 distinct molecular
weights, 68-200 kD, which are the products of distinct genes and can also be
distinguished by immunohistology (Lee et al 1982). Following damage, they
rapidly disappear and are digested by axonal calcium-activated proteases
(Schlaepfer & Hasler 1979). Neurotubules are akin to microtubules in other
cell types and act to give cytoskeletal support.
Axons are unable to synthesise proteins thus are dependent on the neuronal
cell body or the glial cells for their maintenance. Macromolecules are
transported along both directions of the axon concurrently and at various
speeds, from around 1mm to 468mm/day (Tytell et a l 1981). The slow
anterograde transport is for the bulk movement of neurofilaments and is
equivalent to the rate of axonal outgrowth sprouting (Di Giamberardino et al
1973). An important function of retrograde transport, but not the only activity,
is uptake of exogenous growth factors produced by the end organs and
transported back to the neuronal cell body (Ebendal et a l 1980).
Nerve fibres, together with their supporting Schwann cells (SC), form the bulk
of the peripheral nerves. These are surrounded by an outer epineurium,
composed by connective tissue, fibroblasts and blood vessels, a perineurium,
which surrounds the nerve fascicles, and an inner endoneurium. Thus a
diffusion barrier is formed that isolates the endoneurium from the connective
tissue surrounding the epineurium (Ahmed & Weller 1979). The endoneurium
contains the supporting elements of the nerve fibres, which includes SC,
fibroblasts, occasional mast and fat cells and extracellular matrix (ECM)
(Thomas et a l 1993). Although the fascicular pattern of the nerve fibre is
highly variable, there is a high degree of segregation of afferent and efferent
fascicles of pure motor or sensory nerves within each fascicle and nerve type
(Terzis & Smith 1990).
1.1.2 Schwann cells
These are the principal glial cells o f the peripheral nerve, and they were first described by Theodor Schwann (1810-1882) (Figure. 1.1), w ho reported the cell m erely as an incidental passing interest in histology (Figure. 1.2). He described SC as a part o f his general theory o f cellular structure (Causey 1960).
Figure 1.1 Portrait o f T. Schwann
V Y"
Figure 1.2 Original drawings o f T. Schwann (from Causey, 1960)
Schwann was under the impression that these cells developed from the axons
(or Remak fibres as he described them). However, it is now clear that SC
originate from the neural crest and to a lesser extent from the neural tube
(lessen & Mirsky 1998). The irreversible step from neural crest cells to SC
precursors is observed in cells that express high levels of mRNA for PO,
which is a major myelin protein (Mirsky & lessen 1999). The number of cells
migrating from the neural crest to become SC is small compared to the final
number of mature cells in the peripheral nerve. SC precursors do not express
SlOO marker but within days and early during gestation the typical SlOO
antigen is expressed in the cells (lessen & Mirsky 1991). The precursors of
SC undergo a proliferative phase and the exact mechanism that regulates this
proliferation remains unclear. It may be that the developing embryonic
neurons partly regulate this activity as they express high levels of neuregulins,
in particular glial growth factor (GGF) (Shah et al 1994). GGF is a potent SC
mitogen (Marchionni et a l 1993) and it interacts with SC heterodiameric
receptors composed of c-erbB2, c-erbB3 and c-erbB4 (Grinspan et a l 1996).
Deficiency of SC is observed in mice with the deletion of neuregulin-1
(Meyer & Birchmeier 1995). Further evidence on the role of the neurons is
evident when SC isolated from axons become quiescent in vitro. SC mitosis
continues until there is a 1:1 ratio between axons and myelinating SC, and the
segment of axon that is myelinated by a single SC is termed node of Ranvier.
As myelination proceeds, there is a down regulation of GGF receptors, erbB2
and erbB3 (Mirsky & lessen 1999).
There are two very distinct categories of SC, myelinating and non
myelinating. Axons, whether myelinated or unmyelinated are ensheathed by
SC along their entire length. A feature of SC is the deposition of a basal
lamina, a component of extracellular matrix, which surrounds the axons and
isolates them from the surrounding matrix. Axonal contact has been shown to
be a factor for differentiation of SC to myelin forming phenotype (lessen et al
1987). Axonal diameter is also important, as only axons with a diameter larger
than 0.7pm undergo myelination (Windebank et al 1985), yet the identity of
myelination signal remains unclear. Recently, it has been shown that SC
synthesise progesterone, which stimulates myelin formation (Chan et al
2000) and blocking of progesterone receptors also inhibits myelination
(Koenig et a l 2000).
The phenotypic markers expressed by SC seem to be dependent to their
related axon rather than an inherent difference in SC. This is evident as after
loss of axonal contact, SC readily revert back to the expression of markers
characteristic of immature non-myelinating SC (lessen & Richardson 1996)
and this phenomenon is irrespective of SC type. Some of the phenotypic
markers are common to all mature SC, such as SlOO, vimentin and laminin
(Scherer 1997). Other markers like PO, myelin basic protein, myelin
associated glycoprotein are restricted to myelin forming SC (Scherer 1997).
Some markers are exclusive to non-myelinating SC such as intermediate glial
fibrillary acid protein (GFAP), p75 (low affinity nerve growth factor
receptor), neural cell adhesion molecule (NCAM) and Ll (Jessen & Mirsky
1992). Thus it is possible to ascertain the SC phenotype using
immunohistochemical stains for the distinctive phenotypic markers.
1.1.3 Extracellular matrix
Neuronal axons and SC are enmeshed in a complex extracellular matrix
(ECM), whose two major components are basal lamina and fibrillar matrix.
The fibrillar matrix is made up mostly of type I and III collagen, while the
basal lamina contains types IV and V collagen, the glycoproteins laminin,
fibronectin and tenascin, as well as heparan sulphate (Chemousov & Carey
2000). The ECM components act together to provide structural support for the
cellular elements and affect their behaviour during development and maturity.
Tissue culture studies have defined the ECM products that are produced by
SC, how their production is controlled, and how endoneurial and perineural
fibroblasts co-operate and are regulated by SC in organising ECM. Fibroblasts
can also induce synthesis of ECM via SC (Obremski et al. 1993), and in turn
the SC signal to the surrounding connective tissue, possibly via Desert
Hedgehog molecules, to organise perineurial fibroblasts (Mirsky & Jessen
1999). ECM is not produced and organised by neurons cultured alone,
whereas cultured SC can express basal lamina components on their surface,
although these are not formed into basal lamina as identifiable by electron
microscopy (Baron-Van Evercooren et al 1986). It is in co-cultures of
neurons and SC that a basal lamina is formed on the axonal surface of SC, and
a continuous basal lamina production depends on direct contact with axons
(Bunge et a l 1990). Thus SC have a critical role in the production of ECM,
which is essential for the ensheathing of the axons. The importance of ECM
was demonstrated in tissue culture studies where collagen production and
subsequent myelination was prevented in medium without ascorbic acid
(Eldridge et al. 1989). An important effect of the basal lamina is to polarise
the myelinating SC, as the surface free of the basal lamina is responsible for
axonal adhesion (Chemousov & Carey 2000). Laminin and fibronectin are
potent promoters of cell adhesion, while migration, adhesion and signalling
functions of ECM are mediated by integrin receptors on SC (Chemousov &
Carey 2000). Antibodies to p i fragment of the integrin receptor block
migration on laminin 1 and 2 and myelination, while av fragment may be
involved in migration of SC on fibronectin (Mirsky & lessen 1999).
1.2
Axonal degeneration
Axonal degeneration is the default biological response following any distal
axonopathy. There are many causes of axonopathy either hereditary or
acquired and a review is given in Greenfield’s Neuropathology (Graham &
Lantos 1997). The focus of this section is on traumatic axotomy.
1.2.1 Classification of nerve injury
As a results of a large retrospective survey, a scheme for classification of
traumatic nerve injuries was developed by Seddon (1954a) and subsequently
by Sunderland (1978). In first degree injuries (neuropraxia), axonal continuity
is maintained and the decreased function is reversible. In second degree
injuries (axonotmesis), there is a complete axonal dismption but the basal
lamina is intact and recovery occurs in months. In third degree injuries, the
basal lamina is also interrupted and there is a complete functional impairment.
In the fourth and fifth degree injuries (neurotmesis), there is a full structural
disruption and microsurgical intervention can improve the prognosis. Nerve
injuries rarely follow such defined classification except fourth and fifth degree
lesions where all components are damaged. It is this type of injury that has
been studied in the experiments described in this thesis.
1.2.2 Wallerian degeneration
Following nerve injury, complex changes occur throughout the neuron, both
in the proximal and distal nerve segments, extending to the muscle motor end
plates and the sensory receptors. Chromatolysis takes place in the cell body,
manifested as soma swelling, break up of Nissl's granules and nuclear
eccentricity (Sterman & Delannoy 1985). Protein metabolism is altered and
directed towards repair of the cytoskeleton and regeneration of the axons, with
a corresponding decrease in neurotransmitter production. Neuronal cell death
by apoptosis may occur (Ekstrom 1995, Hart et al 2001), and its extent is
dependent on the site and degree of the injury (Ma et a l 2001). A sequence of
events that occur in the distal and to some extent in the proximal stump was
described by Waller (1850) hence it was called Wallerian degeneration.
However, in his original account Waller only described the distal changes of
myelinated axons.
During the first week after injury remodelling occurs along the whole of the
distal segment and to some degree in the proximal stump, up to the first node
of Ranvier. In the myelinated fibres, axons and myelin sheaths break down.
The firagments are phagocytosed partly by SC (Stoll et al. 1989), but
macrophages are mainly involved in myelin degradation and play a crucial
role in the progress of Wallerian degeneration (Hall 1993). SC in the distal
segment undergo de-differentiation by altering their phenotypic marker
expression by exhibiting low affinity nerve growth factor receptor (LNGFR),
p75 antigen (Nikam et a l 1995), and becoming mitotic and proliferate,
peaking at 3 to 5 days post axotomy. Following mitosis there is a longitudinal
alignment of SC within the basal lamina, which results in the formation of
bands of Biingner. This alignment of SC is precursor to regeneration of the
axons, which find a suitable substrate and directional cues for their regrowth
towards and into the distal stump. The changes in unmyelinated fibres are
similar, but longitudinal columns of SC in form seen in the band of Biingner
do not develop (Graham & Lantos 1997).
It is unclear what is the signal initiating Wallerian degeneration, but there is
some evidence that disruption of axonal transport results in SC de-
differentiation (Wu et a l 1994). That axonal degeneration may be a signal for
Wallerian degeneration is also confirmed by studies in C57BL/Wld^ mice,
where delayed axonal degeneration results in postponement of SC
proliferation and macrophage invasion (Glass et a l 1993). Following axonal
degeneration, SC start to produce neuregulins, which induce glial cell mitosis
(Raabe et a l 1996). Thus SC may be partially responsible for their own
proliferation during Wallerian degeneration, probably acting via autocrine or
paracrine mechanisms (Carroll et a l 1997). There is also evidence that
recruited macrophages may stimulate SC proliferation (Kubota & Suzuki
2000).
1.3 Axonal regeneration
Axonal sprouting begins rapidly, starting at 6 hours after the injury. This
happens before any change in the cell body becomes apparent; thus the ability
to grow sprouts may be intrinsic to the axon. The degree of injury largely
dictates the mode of axonal growth. After transection and formation of a nerve
gap, SC and fibroblasts from both the proximal and distal stump migrate into
the gap. There is collateral axonal sprouting originating at the node of Ranvier
immediately proximal to the site of the injury, and as many as 25 sprouts may
stem from a single axon, possibly induced by the Wallerian degeneration
(Slack et al 1979; Graham & Lantos 1997). The growth cones, which are the
tip of the axonal sprouts, extend along the inner surface of SC or through the
band of Biingner, into the distal nerve segment (Martini et a l 1990; Son &
Thompson 1995). Axonal sprouting decreases with time, which is probably
related to the successful contact with the distal target organ (Sanders & Young
1976).
The mechanism of growth cone movement is not fully elucidated. Filopodia at
the tip of the growth cone are actin rich and may sense the gradient of trophic
and adhesive factors (O’Connor et a l 1990). The forward movement may be
only partially explained by the push of slow transport of microtubules as the
forward movements of growth cone and microtubules formation do not fully
correlate (Tanaka & Kirschner 1991). Axonal elongation behind the grov^h
cone needs addition of new membrane and cytoskeleton, and evidence
suggests that the new membrane is added both at the growth cone and along
the length of the axon (Popov et a l 1993). The growth cone is well supplied
by actin and tubulin but deficient in neurofilaments, thus there is a zone of
neurofilament deficient axon behind the growth cone (McQuarrie & Lasek
1989), which may result in slight underestimation of the regeneration distance
when using this marker.
1.3.1 Role of Schwann cells
SC and the associated basal laminae support nerve regeneration (Ide et al
1983; Sketelj et a l 1989; Son & Thompson 1995). SC migration into the site
of injury is independent of the accompanying axon (Anderson et a l 1991) but
the axonal growth is severely impaired when SC mitosis is prevented (Hall
1986a; Bresjanac & Sketelj 1989). The growth cone will preferentially adhere
to denervated SC rather than to their basal lamina (Martini et a l 1994). In
addition, SC may induce axonal sprouting (Son et a l 1996) and play an active
role in supporting neurons during the regeneration process by giving
guidance, providing neurotrophic support and regulating axonal differentiation
(Son & Thompson 1995). Initially SC surround a number of regenerating
axons but with time a 1:1 relationship with larger axons is achieved. Axon-SC
interactions during regeneration are recapitulated in those events also seen
during development (cf. 1.1.2).
Following injury, SC upregulate the synthesis of many known trophic factors,
including nerve growth factor (NFG) (Heumann et a l 1987), brain-derived
neurotrophic factor (BDNF) (Acheson et al 1991), platelet-derived growth
factor (PDGF) (Eccleston et a l 1993), insulin-like growth factor (Cheng et al
1996), ciliary neurotrophic factor (CNTF) (Friedman et a l 1992) and
leukaemia inhibitory factor (LIF) (Matsuoka et a l 1997), all of which play a
role in nerve regeneration (Terenghi 1999). These growth factors can promote
regeneration by providing neurotrophic support while other molecules
synthesised by the SC can contribute to axonal guidance.
SC express a number of cell adhesion molecules such as NCAM, LI and
laminin, which are upregulated following denervation and by two weeks after
injury SC display LI and NCAM phenotype (Martini 1994). The presence of
LI and NCAM on SC is essential for adequate axonal regrowth as they
mediate SC interaction with counterpart molecules present on the growth cone
(lessen & Richardson 1996). Therefore in summary, SC give both structural
and trophic guidance to the regenerating neurons with their axons during this
process.
1.4
Peripheral nerve repair
Currently, primary epineurial repair is used where there is no loss of nerve
tissue (Bora et a l 1976). Reconstruction is a bigger challenge when there is a
gap in the nerve and the ends cannot be joined directly. The focus of this
thesis is on such situations when gap repair is carried out.
For many centuries surgeons avoided touching the nerve stumps for the fear of
causing convulsions despite the attempts by Paul of Aegina (625-690) to
approximate nerve stumps before closing the wound. The Persian physician
Avicenna (980-1037) achieved indirect closure of nerve ends by
approximating the remaining soft tissue (Omer et al 1997). The first use of
nerve transplant in human to bridge a gap was reported in 1878 by Albert
(Davis & Cleveland 1934). However, the first successful use of nerve graft
was described by Bunnell (1927). At the time this procedure had a mixed
reception, with some strong opposition (Platt 1919; Stopford 1920). However,
following publication of a successful series by Huber (1919) and the wide use
with this technique during World War II (Seddon 1954b), nerve grafting was
accepted as a standard method of nerve gap repair. Since then the results of
nerve gap repair have significantly improved, especially following
popularisation of the use of the microscope during surgery (Millesi 1973;
Stancic et al. 1998). Yet, despite advances in microsurgical techniques and
instrumentation, the functional recovery following nerve injury has remained
unsatisfactory (Mackinnon & Dellon 1988). Full functional recovery in
peripheral nerve reconstruction is still elusive and it is generally agreed that
further improvement in outcome will not come fi*om further development in
surgical techniques. It is becoming clear that a complete understanding of the
pathophysiological events of degeneration and regeneration is needed, in order
to manipulate correctly the microenvironment of the nerve injury and to
obtain optimal nerve regeneration.
1.4.1 Nerve gap reconstruction
1.4.1.1 Nerve graft
Nerve grafting is commonly performed as a non-vascularized tissue transfer,
while vascularized nerve grafts are generally performed when there is an
associated vascular deficit or injury (Townsend & Taylor 1984). For nerve
autograft, usually a cutaneous sensory nerve such as the sural nerve, is used to
bridge the nerve gap. The recovery following nerve grafting depends on a
number of factors including the type of the nerve involved, delay at the time
of repair, associated morbidities, soft tissue bed, tension of repair, and the age
of the patient (Vanderhooft 2000). Generally the results of nerve graft in the
upper limb are better than those in the lower limb (Donzelli et al. 1998).
However, the results of a large series by MacKinnon and Dellon has
demonstrated that full functional recovery is never achieved (Mackinnon &
Dellon 1988).
The harvest of a nerve graft results in co-morbidity for the patient, including a
further wound site, loss of sensation and possible formation of painful
neuroma at the donor site. The harvested nerve is often of a smaller calibre
and there are a limited number of nerves that can be sacrificed for this
purpose. In order to overcome the above adversities, allografts have been tried
with some success (Berger & Lassner 1994), the limiting factor remains the
difficulties associated with immunosuppression (Matsuyama et al. 2000).
Xenografts have also been tried and shown to be inferior to autograft (Accioli,
V et al 1999). There has been increasing interest in developing a different
method to nerve grafting, and recently there has been renewed interest in
to side neurorrhaphy (Zhang et a l 2000) but success remains unproven (al-
Qattan & al-Thunyan 1998; Rowan et al 2000). Nerve expansion technique
has been reported to have deleterious effects on nerve function (Milner &
Wilkins 1992). Whilst other ingenious methods have been described (Ryoke
et a l 2000; Ayhan et a l 2000), autologous free nerve graft remains the
standard method for clinical nerve gap repair.
1.4.1.2 Conduits and other alternatives methods
The search for alternative methods to bridge a gap in an injured peripheral
nerve is nothing new and has encompassed the use of both autologous as well
as artificial and biodegradable materials. In 1880 Gluck used surgical drains
of bone to repair nerve and eleven years later, Biingner used a segment of
artery to repair the hypoglossal nerve of a dog with some success (Gliick
1880; Hunger 1891). Stopford and Platt reported poor results in their series of
experiments using veins for grafting (Platt 1919; Stopford 1920) and the
interest in conduits waned until a report by Weiss re-kindled interest in the
field (Weiss & Taylor 1946). For structural reasons blood vessels were an
obvious choice and they were first used to protect nerve anastomosis (Miglets
& Thomas 1973) but also as a conduit (Walton et a l 1989) with a number of
modifications (Benito-Ruiz et a l 1994; Ferrari et a l 1999). The results were
still inferior to nerve grafts and the technique has mainly been restricted for
the repair of short gaps (Chiu 1999). Muscle grafts attracted interest as the
basal lamina of the myofibrils was similar to the structure of nerve grafts
(Fawcett & Keynes 1986) and could guide regenerating axons (Davies et al
1987). It was shown that pre-degeneration of the muscle graft (Glasby et al
1986; Hall 1997) was necessary and that the method of pre-degeneration was
important (Whitworth et a l 1995a). Further improvements on regeneration
were seen when nerve and muscle were combined as a sandwich graft (Calder
& Green 1995; Whitworth et a l 1995b). Encouraging results have also been
reported using the combination of veins filled with muscle grafts (Battiston et
al 2000), However, the overall results of the previous methods are still not
satisfactory especially for long nerve defects (Hems & Glasby 1993).
The use of non-biological materials for gap repair was not introduced until
one century after trials of biological materials and a number of materials have
been used for making conduits: magnesium, formalin treated casein, gelatin,
rubber, Millipore and cellulose (Fields et a l 1989), but all with no success.
This resulted that Sundeiiand concluded the use of non-biological materials
had no value in nerve gap repair and could even worsen the regeneration by
introducing infections and fibrosis (Sunderland 1978). Interest in non-
biological conduits was re-started when Lundborg showed regeneration across
a silicone conduit (Lundborg et al 1982), however, their removal after
regeneration was recommended because of their rigidity causing chronic
irritation of nerve (Lundborg et a l 1994). The most promising conduits have
been biodegradable, manufactured from naturally occurring or synthetic
substances (Fields et a l 1989; Doolabh et al 1996; Strauch 2000). Advances
in material engineering and improved production techniques has also enabled
surface texture modification which has shown to be important in guiding
cellular migration (Curtis & Wilkinson 1997; Curtis & Wilkinson 2001) and
regenerating axons (Guenard et a l 1991).
A number of biodegradable conduits have been shown to be useful in the
nerve gap repair: such as fibronectin, precipitated from blood and shaped into
mats (Whitworth et al. 1995c), collagen (Madorsky et al. 1998), polylactic
acid (Evans et al. 1999), polyglycolic acid (Matsumoto et al. 2000), their co
polymer (Hadlock et al. 1999), polyester urethane (Borkenhagen et al. 1998)
as well as inorganic materials (Gilchrist et al. 1998).
Poly-3-hydroxybutyrate (PHB) is an example of a biodegradable polymer of
natural origin that can be easily produced in vitro. PHB granules are a natural
storage product of bacteria and can be isolated by solvent extraction. PHB is
non-toxic, biocompatible, non-antigenic and resorbable by hydrolytic
degradation (Holmes 1988) and it has been made into sheets which have
longitudinally aligned fibres, ideal for contact guidance of neuronal and glial
components. Nerve conduits made from PHB sheets have been used
effectively for primary nerve repair (Hazari et al. 1999a), as well as being
useful for nerve gap repair (Hazari et al. 1999b; Young et al. 2001). Its use
has not produced excessive fibrosis or inflammatory response (Hazari et al.
1999a) and PHB sheets have also been used clinically in cardiovascular
surgery (Duvemoy er a/. 1995).
Although the PHB conduit may give directional guidance to the sprouting
axons through its microgeometry (Aebischer et al. 1990), there is evidently a
lack of supporting elements within the conduit necessary for bridging long
nerve gaps (Young et al. 2001). The need of supporting elements for nerve
regeneration is apparent from experiments where a nerve segment has been
inserted into a muscle graft, as a source of autologous SC that could help and
improve the regenerative process of the axons (Calder & Green 1995;
Whitworth er a/. 1995a).
However, recovery following use of conduits is inferior to that using
autologous nerve grafts. Even the use of nerve grafts for the peripheral nerve
reconstruction does not result in full functional recovery. Furthermore, their
use necessitates sacrifice of normal nerve, such as sural nerve, and results in
extra morbidity (Mackinnon & Dellon 1988). Hence there is a clinical need to
improve the outcome of the conduit repair to be used as an alternative the
nerve graft.
1.4.2 Time factor
In most experimental studies, nerve gap repair is carried out immediately after
injury, which is optimal but in reality there is often a delay affecting neuronal
survival and causing atrophy of the distal nerve and end organs. Following
axotomy, SC survive in the distal stump for some time (Meier et al. 1999), but
there is a gradual dispersion and disappearance of SC basement membrane
(Gianni & Dyck 1993). Furthermore, the ability of SC to respond to axonal
mitogens reduces by time (Li et al 1997) and the terminal SC, teloglia,
undergo apoptosis if regeneration does not occur within a certain period
(Trachtenberg & Thompson 1996).
Due to the continuing collagen deposition, failure in regeneration results in
irreversible shrinkage of the distal endoneurial sheath (Thomas 1964) and two
years after injury, the cross-sectional area is 1% of the normal size (Terzis &
Smith 1990). This causes scar formation and a physical block for regenerating
axons resulting in neuroma formation. The deterioration in SC and the distal
nerve fibrosis together with the target organ deterioration (Fu S.Y. & Gordon
T. 1995) may explain the deleterious effect of delay on regeneration.
1.5
Bioengineered nerve conduits
Biocompatible conduits may be combined with cultured cells and/or growth
factors. Hence, the requirements of regeneration can be met by concentrating
within the conduit all the elements needed for a successful axonal regrowth
(Heath & Rutkowski 1998; Hudson et al. 2000; Schachner 2000), in essence
producing a bioengineered nerve conduit.
It is clear that SC within such a nerve play a crucial role, however, several
other elements need to be taken into consideration such as the matrix in which
SC are suspended as ECM and perineurial fibroblasts. The loss of
neurotrophic factors produced fi*om end organs may be compensated by the
addition of extra growth factors, an alternative strategy would be to boost the
synthesis of growth factors by transplanted SC through genetic engineering
techniques.
1.5.1 Transplantation of cultured Schwann cells
SC are important for neuronal survival and development of axonal
characteristics (lessen & Mirsky 1999), and are essential for axonal regrowth
following injury (Hall 1986b). SC transplantation has been shown to enhance
both peripheral and central nerve regeneration (Smith & Stevenson 1988;
Guenard et a l 1992; Levi et a l 1994; Guest et a l 1997). Before this treatment
can be used clinically, further improvements of the effects of SC therapy
should be demonstrated. SC are quiescent in vitro and do not divide
spontaneously. Over the last decade SC culture techniques have been
improved (Rutkowski et a l 1995; Casella et a l 1996; Abrams et a l 1998;
Lopez & De 1999) thanks also to the availability of the SC mitogen, GGF II
(Marchionni et a l 1993). Consequently, it has been possible to obtain a large
number of purified cultured primary SC from a small nerve biopsy. It has
been demonstrated that, at least in short term, cultured SC maintain their
phenotype (Rutkowski et a l 1990) thus may be suitable for implantation.
The use of SC for peripheral nerve repair is not a new concept. However,
before human trials of this therapeutic modality may be contemplated, a
number of questions need to be answered. Firstly, the safety of SC
transplantation need to be demonstrated, such as undergoing metaplastic or
neoplastic changes. Additionally, active participation of transplanted SC in the
regenerative process should be demonstrated. These two questions may be
addressed by a reliable method of tracking SC following transplantation.
Similarly, a protocol needs to be devised such that conversion from the
experimental model to the clinical setting would be technically and ethically
feasible and acceptable. This entails use of materials and matrices that may be
of use in the clinical setting. A further question that needs addressing is the
optimal number of transplanted SC that may be used to give the optimal
results. This would not only achieve acceptable functional recovery but also
avoid wastage of a highly intensive procedure, which would be crucial in its
uptake in routine clinical practice. Unfortunately, previous papers have left the
above questions unanswered (cf. Appendix 4).
SC identification has been poorly addressed and some investigators have not
attempted to identify the SC following transplantation (Brown et al. 1996;
Bryan et al. 1996), whilst others have used either chemical labelling (Kim et
al. 1994; Ansselin et al. 1997; Rodriguez et al. 2000a) or genetic labelling
(Aguayo et al. 1977). However, the effect of the labelling techniques on the
integrity of SC is not clear. Despite the numerous variables and inconsistent
methodologies, previous studies agree that transplanted SC improve
peripheral nerve regeneration, however, the characterisation of an effective
methodology is still forthcoming.
It is possible that the time delay prior to obtaining an adequate number of
autologous SC for transplantation as well as the local availability of expertise
would limit the clinical use of SC transplantation for peripheral nerve
reconstruction. Allogeneic SC transplantation is therefore an attractive option
as it would allow using an off-the-shelf population of screened and cultured
SC ready for transplantation. Transplantation of allogeneic SC without
immunosuppression has been reported without success (Guenard et al. 1992)
although their participation in the regeneration process was not evident and a
clear identification of transplanted SC was not addressed and it may be
possible that rejection occurred before regeneration.
1.5.1.1 Schwann cell chemical labelling
In order to investigate the cellular mechanism and contribution of transplanted
SC for nerve regeneration, reliable markers are needed to distinguish donor
and host SC during the regeneration process in vivo. This is especially
important in peripheral nerves, as SC are recruited equally fi*om the damaged
proximal and distal ends (Bardosi 1989)(Diaz-Flores et al. 1995). The
labelling technique needs to be stable and uniform, but should not affect the
cell viability, morphology and characteristics of the cells.
Radioactive probes such as Iodine and Chromium have been used for
lymphocytic cell tracking, however, toxicity, internal radiation effects, poor
uptake and rapid elution (Horan 1990) make these methodologies unsuitable
for long term in vivo transplantation. Fluorescent probes such as fluorescein
and rhodamine isothiocyanate rapidly escape during the first 5 hours of
tracking (Olszewski 1987) making them incompatible for use in SC. Dyes
such as indocarbocyanine have been shown to suffer from non-uniform
labelling when used in neuronal cells (Honig 1986).
Two reporter molecules were identified in this study, which were considered
as possibly useful for a more permanent and efficient labelling of SC.
Bisbenzimide (Hoechst 33342) has been suggested as a suitable marker for SC
prior to transplantation (Baron-Van Evercooren et al 1991) and has been used
in vivo (Casella et a l 1996; Ansselin et al 1997; Hermanns et al 1997).
H33342 reversibly binds to adenine-thymidine rich regions of chromatin and
is rapidly incorporated into the SC transplantation. PKH26 is a fluorescent
dye that has been claimed to have superior overall profile for use in cell
trafficking studies (Horan 1998) and has also been used for SC labelling in
vivo (Hermanns et al 1997). PKH26 is a highly aliphatic reporter molecule
containing a fiuorochrome, which is entrapped after incorporation into the cell
membrane due to its differential solubility in an aqueous-lipid environment.
Notwithstanding these recent uses, the effect of labelling with H33342 and
PKH26 on SC has not been adequately documented, and it was investigated in
this study.
1.5.1.2 Schwa fin cell genetic labelling
Genetic markers may be used as an alternative to chemical. A replication
deficient retroviral vector may be used to permanently introduce a marker
gene such as lacZ into SC (Bunge et a l 1989; Ferry et a l 1991). Similar
methodology has successfully been utilised in our centre for the identification
of transplanted cultured kératinocytes for dermal replacement (Bevan et al
1997; Ng era/. 1997).
A further advantage is that the safety and efficacy of the genetic engineering
method utilised may be characterised by the introduction of a gene such as
lacZ gene that does not have products which would interact with the nerve
regeneration. The characterisation of SC would involve ensuring that the
proliferation and viability of cells were not affected following transduction.
Additionally, it is important to demonstrate that following transduction SC
preserve their chromosomal configuration by maintaining the Hayflick limit
(Hayflick & Moorhead 1961) thus they do not become immortalised. Finally,
SC phenotypic expression of markers should be preserved.
1.5.2 Matrix
Transplantation matrix should permit cell proliferation, without toxicity to
either SC or neurons, while lasting long enough to entrap the cells in the
conduit for the duration of axonal regeneration. It should also allow and
support penetration of regenerating axons, being sufficiently porous to mimic
the function of natural extracellular matrix in encouraging tissue formation
from the transplanted cell (Smidsord O & Skjak-Braek 1990). In addition, the
scaffold must withstand in vitro handling as well as surgical procedures
necessary for implantation.
Despite the interest shown by investigators in the concept of nerve guidance,
little attention has been given to find a suitable carrying matrix for growth
factors or cultured cells. Growth factors have usually been applied to the
nerve in physiological solutions, with a limited availability and relatively
short degradation profile (Rich et a l 1989). Bioactive fibronectin mat has
been used for delivery of growth factors to improve nerve regeneration
(Whitworth et a l 1996; Sterne et al 1997; Ahmed et a l 1999), and a number
of cell matrices have been used for SC transplantation (Guenard et a l 1992;
Kim et a l 1994; Silverman et al 1999; Solchaga et a l 1999). Recently,
hyaluronic acid has been modified and made in to strands, which enhanced
ability to allow SC attachment (Hu et al 2000) and magnetically aligned
collagen gel filling has been reported to improve regeneration (Ceballos et al
1999). However, collagen, fibrin, hyaluronic acid are of animal origin and
their source may represent a problem for their clinical application. Also these
matrices maybe resorbed or broken down before adequate axonal regeneration
and maturation has taken place, hence the transplanted SC would lose its
extracellular supporting framework.
Matrigel has been used as a SC delivery matrix (Rodriguez et a l 2000b) but it
is extracted fi*om sarcoma cell lines and may not be suitable to be used in
human. Patterning technology has been shown to allow modification of
matrices to give direction to the nerve regeneration (Patel et a l 1998).
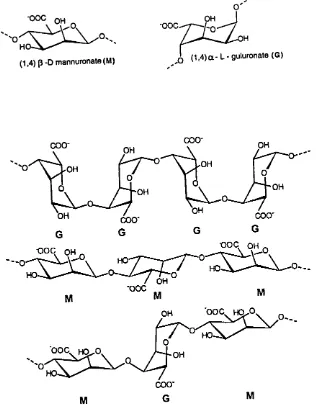
1.5.2.1 Alginate
Alginate hydrogel is a naturally derived water-soluble polymer extracted firom
brown seaweed algae (Figure 1.3). It is available as an ultra-pure, endotoxin
free form. Alginate hydrogel is a linear co-polymer composed of a varying
proportion of 1, 4-linked p-D-mannuronate (M) and 1, 4-linked a-L-
glucuronate (G) blocks, which depends on the algae of origin (Figure 1.4).
The viscosity of its solution is dependent on the length of the polymer, longer
units being more viscous. Biocompatibility of high mannuronate residue
alginate has been demonstrated both in vitro and in vivo (Klock et a l 1997).
Also low viscosity mannuronate (LVM) alginate has been demonstrated to be
more amenable to molecular difftision (Amsden & Turner 1999), which is
important for potential use as a cell suspension matrix.
Alginate extraction
Alginic acid
Depyrogenisation
Micro and ultrafiltration
Figure 1.3 Extraction process of alginate from seaweed. Blue: crude production, green: purification process. (Adapted from
-o o c OH
" O
(1,4) (3 -D mannuronate (M) Q (1,4)a - L - guluronate (G)
COO" COO-OH OH •OH OH OH OH 000' G G G COO'
• o o c OH OOC OH
HO HO. HO-OH M 'OOC M OH HO-OOC h q^o,
OH
COO-M M
Figure 1.4 Molecular structure of alginate G blocks, glucuronate, M
block mannuronate (Adapted from Pronova literature, Norway)
Mammalian cells lack enzymes to digest alginates but as calcium ions
dissolve out of alginate hydrogel, the gel structure is lost and is slowly
excreted renally. Alginate hydrogel has been extensively used in
microencapsulation of pancreatic islet cells for transplantation and treatment
of diabetes mellitus. It has also been used to implant neuronal cells (Boisseau
et al. 1993), genetically modified fibroblasts secreting nerve growth factor
(Maysinger et a l 1994) and for controlled-release of various neurotrophic
factors in central (Maysinger et al 1996) as well as in peripheral nervous
system (Suzuki et al 1999). Furthermore, rods made from alginate hydrogel
and loaded with leukaemic inhibitory factor (LIF) have been reported to give a
sustained release and enhance nerve regeneration (Austin et a l 1997).
Potentially alginate hydrogel may be useful as a matrix for SC suspension,
although it is not clear how SC would react to this matrix. LVM alginate
appears to be useful for SC suspension. Also, alginate hydrogel forms gel
through chemical crosslinkage with multivalent cations e.g. calcium. Hence it
is important to ensure that the exposure to higher calcium concentration
needed for setting of alginate hydrogel does not have deleterious effects on SC
viability and axonal regeneration.
1.5.3 Role of gene therapy in nerve repair
The purpose of gene therapy is to transfer DNA (for example of growth
factors) into the cells such that DNA is synthesised in these cells and
recombinant protein expressed (Buti et a l 1996). Two main methods are viral
mediated transfer, such as retroviral vector (Robbins et a l 1998) and non-viral
techniques, such as plasmid-mediated DNA transfer (Brown et al 1981). It
may also be possible to increase the availability of neurotrophic factors by the
introduction of their DNA. This may be done with either an ex-vivo or an in