Computational Analysis of Functional Sites in Proteins
Full text
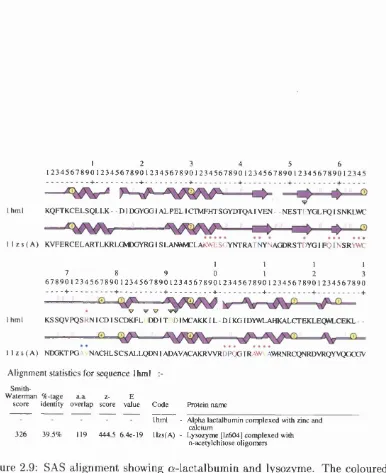
Figure
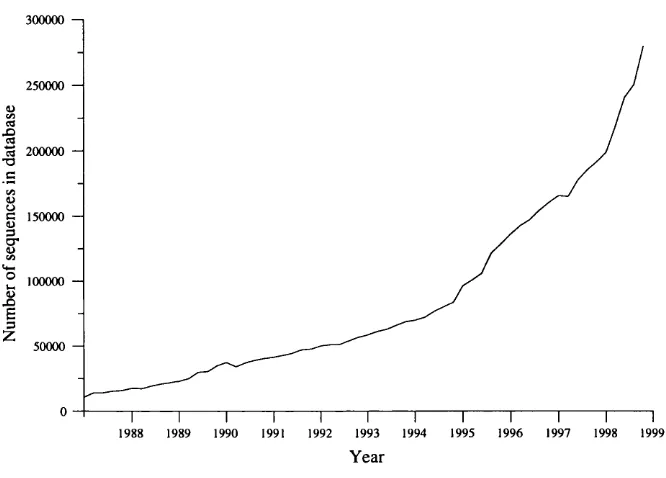
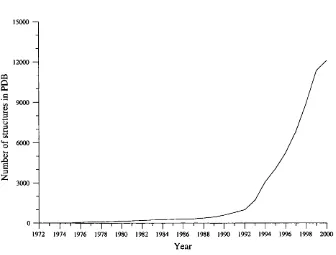
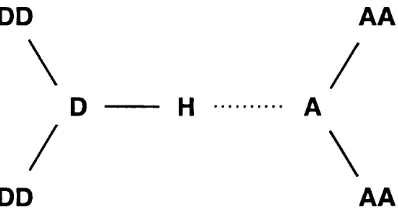
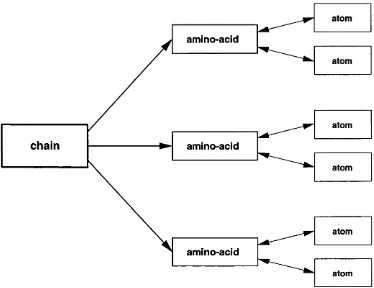
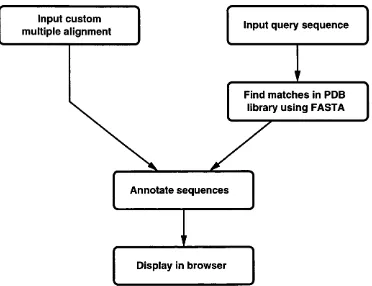
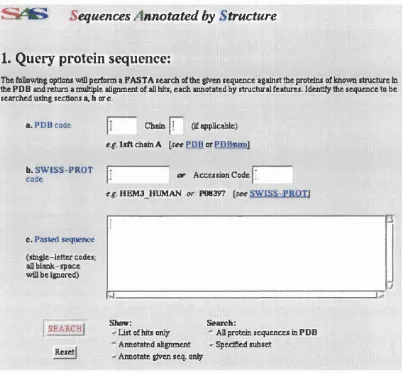

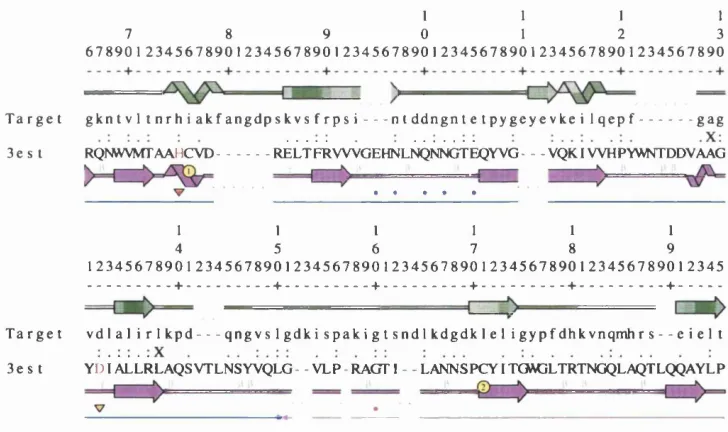
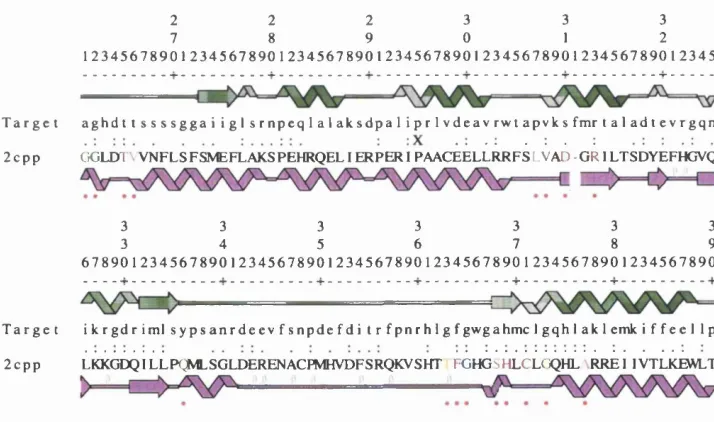
Related documents
Field experiments were conducted at Ebonyi State University Research Farm during 2009 and 2010 farming seasons to evaluate the effect of intercropping maize with
Collaborative Assessment and Management of Suicidality training: The effect on the knowledge, skills, and attitudes of mental health professionals and trainees. Dissertation
To access IPv4 and IPv6 settings in CentreWare Internet Services, enable TCP/IP at the printer control panel.. If you disable TCP/IP or change the IP address, any dependent
Benchmark between the security of NoSQL database and read–write perfor- mance of queries using both the schema suggested by the proposed model and the schema produced using
Keywords:- Cement composites, concrete, pozzolan, rice husk ash, sandcrete, saw dust ash, soilcrete, ternary blended cement, water-cement
The primary outcomes are between group differences in self-efficacy, measured using the Pain Self Efficacy Questionnaire (PSEQ) and disability measured using the Roland
AIRWAYS ICPs: integrated care pathways for airway diseases; ARIA: Allergic Rhinitis and its Impact on Asthma; COPD: chronic obstructive pulmonary disease; DG: Directorate General;
