University of Pennsylvania
ScholarlyCommons
Publicly Accessible Penn Dissertations
1-1-2014
Novel Insights into the Allosteric Activation of the
Epidermal Growth Factor Receptor
Nicholas Bessman
University of Pennsylvania, nbessman1@gmail.com
Follow this and additional works at:
http://repository.upenn.edu/edissertations
Part of the
Biochemistry Commons
This paper is posted at ScholarlyCommons.http://repository.upenn.edu/edissertations/1207
For more information, please contactlibraryrepository@pobox.upenn.edu.
Recommended Citation
Bessman, Nicholas, "Novel Insights into the Allosteric Activation of the Epidermal Growth Factor Receptor" (2014).Publicly Accessible Penn Dissertations. 1207.
Novel Insights into the Allosteric Activation of the Epidermal Growth
Factor Receptor
Abstract
EGF receptor activation requires both ligand-binding and receptor-mediated dimerization through receptor
domain II. The relationship between these processes, however, remains unclear. We have decoupled these
processes to examine the ligand-binding affinity and the structure of constitutively-monomeric and -dimeric
forms of the EGF receptor, as well as EGF receptor that dimerizes upon ligand-binding. Surprisingly,
monomeric receptor binds to the ligands EGF and TGFα with an affinity equivalent to that of dimerizing
receptor but with a unique binding enthalpy. This shows that monomeric, ligated EGF receptor adopts a state
that is distinct from that of EGF receptor within a homodimer, and this state may be relevant to heterodimeric
ErbB signaling complexes. Constitutively-dimerized receptor binds ligand with elevated affinity; however, it
still requires ligand to form the receptor domain II dimeric interface. In the absence of ligand, no ordered,
receptor domain II-mediated dimer interface is formed. Thus, the affinity effect does not arise from any
pre-organization or stabilization of the ligand-binding sites on the receptor, but rather through an entropic effect
of enforcing dimerization. Thus, pre-formed human receptor dimers require allosteric activation by ligand in
order to signal, and this allosteric mechanism is distinct from that we recently observed for the D.
melanogaster EGF receptor. Our observations on the allosteric mechanism of EGF receptor activation
prompted us to ask whether other EGF receptor ligands may exert unique allosteric effects. To this end, we
investigated the allosteric effects of the ligands Amphiregulin, Epiregulin, and Epigen on EGF receptor. We
report that Epiregulin and Epigen, in particular, exert unique allosteric regulation on the receptor, as
evidenced by divergent effects of EGFR variants on ligand-binding. Finally, we have studied ligand-binding
and dimerization of receptors bearing activating extracellular mutations that cause glioblastoma. We report
that these mutations elevate ligand-binding affinity, but they do not drive receptor dimerization. Our findings
inform a revised model of ligand-induced receptor activation, in which the dimerization interface is highly
sensitive to the presence and the identity of the bound ligand, and the domain I/domain II interface plays a
crucial auto-inhibitory role.
Degree Type
Dissertation
Degree Name
Doctor of Philosophy (PhD)
Graduate Group
Biochemistry & Molecular Biophysics
First Advisor
Mark A. Lemmon
Subject Categories
Biochemistry
NOVEL INSIGHTS IN THE ALLOSTERIC ACTIVATION OF THE EPIDERMAL GROWTH
FACTOR RECEPTOR
Nicholas J. Bessman
A DISSERTATION
in
Biochemistry and Molecular Biophysics
Presented to the Faculties of the University of Pennsylvania
in
Partial Fulfillment of the Requirements for the
Degree of Doctor of Philosophy
2014
Supervisor of Dissertation
_________________________
Mark A. Lemmon
Professor and Chair of Biochemistry and Biophysics
Graduate Group Chairperson
______ ___________
Kathryn M. Ferguson, Associate Professor of Physiology
Dissertation Committee
Kathryn M. Ferguson, Associate Professor of Physiology
Mark I. Greene, Professor of Pathology and Laboratory Medicine
James Shorter, Associate Professor of Biochemistry and Biophysics
Yair Argon, Professor of Pathology and Laboratory Medicine
Gregory D. Van Duyne, Professor of Biochemistry and Biophysics
ii
ACKNOWLEDGMENT
I am grateful to many individuals and institutions for crucial support and encouragement during
my dissertation research. My advisor, Mark Lemmon, provided wisdom and direction throughout.
He provided me with the resources and the freedom to pursue novel and controversial ideas, and
was always excited to discuss the implications of surprising experimental results.
Members of the Lemmon and Ferguson laboratories have further supported my work. I would
particularly like to thank Diego Alvarado, whose work sparked my thesis research; Fumin Shi,
who helped orient me as a new student in the laboratory; Jeannine Mendrola, who managed the
lab throughout my research; Pamela Burgess-Jones, who provided crucial technical and
administrative support; and Atrish Bagchi, with whom I collaborated in studying glioblastoma
variants of EGFR. Outside of these labs, I wish to acknowledge Dewight Williams, who assisted
me with electron microscopic analyses; Kushol Gupta, who provided technical advice for SAXS
and AUC experiments; Steve Stayrook, for technical assistance with X-ray and SAXS
experiments; and Ruth Keris, Lisa Ward, and Angie Young, who provided crucial administrative
support.
Finally, I must express my gratitude for my parents, Carl and Jean Bessman, and for my wife,
Lauren Naliboff, whose loving support throughout the years has always inspired me to pursue my
iii
ABSTRACT
NOVEL INSIGHTS INTO THE ALLOSTERIC ACTIVATION OF THE EPIDERMAL GROWTH
FACTOR RECEPTOR
Nicholas J. Bessman
Mark A. Lemmon
EGF receptor activation requires both ligand-binding and receptor-mediated dimerization through
receptor domain II. The relationship between these processes, however, remains unclear. We
have decoupled these processes to examine the ligand-binding affinity and the structure of
constitutively-monomeric and -dimeric forms of the EGF receptor, as well as EGF receptor that
dimerizes upon ligand-binding. Surprisingly, monomeric receptor binds to the ligands EGF and
TGFα with an affinity equivalent to that of dimerizing receptor but with a unique binding enthalpy.
This shows that monomeric, ligated EGF receptor adopts a state that is distinct from that of EGF
receptor within a homodimer, and this state may be relevant to heterodimeric ErbB signaling
complexes. Constitutively-dimerized receptor binds ligand with elevated affinity; however, it still
requires ligand to form the receptor domain II dimeric interface. In the absence of ligand, no
ordered, receptor domain II-mediated dimer interface is formed. Thus, the affinity effect does not
arise from any pre-organization or stabilization of the ligand-binding sites on the receptor, but
rather through an entropic effect of enforcing dimerization. Thus, pre-formed human receptor
dimers require allosteric activation by ligand in order to signal, and this allosteric mechanism is
distinct from that we recently observed for the D. melanogaster EGF receptor. Our observations
on the allosteric mechanism of EGF receptor activation prompted us to ask whether other EGF
receptor ligands may exert unique allosteric effects. To this end, we investigated the allosteric
effects of the ligands Amphiregulin, Epiregulin, and Epigen on EGF receptor. We report that
Epiregulin and Epigen, in particular, exert unique allosteric regulation on the receptor, as
evidenced by divergent effects of EGFR variants on ligand-binding. Finally, we have studied
iv
glioblastoma. We report that these mutations elevate ligand-binding affinity, but they do not drive
receptor dimerization. Our findings inform a revised model of ligand-induced receptor activation,
in which the dimerization interface is highly sensitive to the presence and the identity of the bound
v
TABLE OF CONTENTS
ACKNOWLEDGMENT
... II
ABSTRACT
... III
LIST OF TABLES
... VII
LIST OF ILLUSTRATIONS
... VIII
CHAPTER 1: EGF RECEPTOR SIGNALING, FROM ORGANISMAL BIOLOGY
TO ATOMIC RESOLUTION
... 1
Receptor tyrosine kinases: a conserved transmembrane signaling module ... 2
Biology of ErbB family RTKs ... 13
Mechanistic insight into ErbB signaling ... 15
CHAPTER 2: PROBING THE RELATIONSHIP BETWEEN LIGAND-BINDING
AND EGF RECEPTOR DIMERIZATION AND ACTIVATION
... 21
Pre-dimerized EGFR requires ligand to form a domain II-mediated dimer... 22
EGFR dimerization does not stabilize ligand-binding ... 27
Pre-dimerized EGFR binds ligand with a high affinity driven by entropy ... 33
Pre-dimerized EGFR exhibits two distinct binding sites in a temperature-dependent manner ... 35
Domain II:Domain IV intramolecular interactions stabilize inactive EGFR enthalpically ... 39
Transforming mutations in the EGFR ECR do not drive receptor dimerization ... 42
Conclusions ... 46
CHAPTER 3: CHARACTERIZING SIGNALING BY DIVERSE EGF RECEPTOR
LIGANDS
... 54
vi
Epigen, Epiregulin, and Amphiregulin bind to EGF receptor with unique thermodynamic
profiles ... 60
Epigen- and Epiregulin-binding exhibit unique coupling to EGFR functional variants ... 63
Conclusions ... 67
CHAPTER 4: PERSPECTIVES AND FUTURE DIRECTIONS
... 70
CHAPTER 5: MATERIALS AND METHODS
... 74
vii
List of Tables
Table 1: Thermodynamics of ligand-binding to dimerizing and monomeric sEGFR…….25
Table 2: Thermodynamics of ligand-binding to ligand-independent sEGFR dimers……31
Table 3: Thermodynamics of ligand-binding to sEGFR bearing compromised ‘tether’
interfaces………..36
Table 4: Thermodynamics of ligand-binding to sEGFR bearing extracellular glioblastoma
driver mutations………40
viii
List of Illustrations
Figure 1: General domain architecture of receptor tyrosine kinases………3
Figure 2: Ligand-induced dimerization activates receptor tyrosine kinases………8
Figure 3: A structure-based model for ligand-induced dimerization and activation
of the EGF receptor………17
Figure 4: EM- and SAXS-derived model for the domain architecture of
ligand-independent sEGFR dimers……….20
Figure 5: EM imaging of ligand-independent sEGFR dimers.……….22
Figure 6: Ligand-binding to monomeric and dimerizing sEGFR……….25
Figure 7: Thermodynamics of receptor dimerization………27
Figure 8: Ligand-binding analysis of ligand-independent sEGFR dimers……….30
Figure 9: Temperature-dependent two-site binding behavior for sEGFR-Fc.…..32
Figure 10: Domain II-mediated dimerization plays a ligand-specific role within
sEGFR-Fc………34
Figure 11: Ligand-binding to sEGFR bearing a compromised tether interface…37
Figure 12: Glioblastoma sEGFR variants favor ligand-binding, but not
dimerization……….40
Figure 13: An updated allosteric model for ligand-induce dimerization of the EGF
receptor………44
Figure 14: Production of recombinant, bio-active low-affinity ligands of the EGF
receptor………54
Figure 15: Thermodynamics of low-affinity ligand binding to sEGFR-Fc………..58
1
2
Receptor tyrosine kinases: a conserved transmembrane signaling module
Receptor tyrosine kinases, or RTKs, are a conserved family of transmembrane signaling proteins
in metazoan organisms. RTKs play a central role in organismal development and patterning.
Beyond development, RTKs serve to maintain physiological homeostasis in mature organisms.
Many of the earliest biochemical paradigms in physiology and development were, many decades
later, found to depend directly upon the transmembrane signaling properties of RTKs. For
example, insulin was initially purified in the early 1920s by Frederick Banting and colleagues, and
it was immediately shown to cure diabetes when injected at regular intervals(Banting, 1922).
Later, Rita Levi-Montalcini and Stanley Cohen, working in close proximity in the 1950s,
discovered two molecules with remarkable stimulatory effects on nerve cells and mouse
development, respectively(Cohen, 1962; Levi-Montalcini, 1960). These two molecules were
termed ‘growth factors’. With the advent of recombinant DNA technology and gene cloning in the
1970s and 1980s, it was recognized that the receptors mediating the biological effects of insulin
and other growth factors constitute just a handful of examples from a broad and conserved family
of RTKs. Each RTK senses a unique set of extracellular ligand molecules and transmits a signal
to the interior of the cell by a broadly conserved enzymatic mechanism (Fig. 1). Accordingly,
RTKs broadly allow individual cells within a multi-cellular organism to respond appropriately to
diverse developmental, physiological, and environmental cues, by exploiting a modular
3
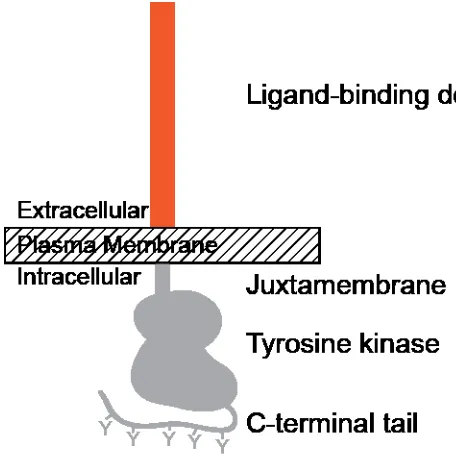
Figure 1: RTKs share a common architecture. In the extracellular regions (orange), a wide
variety of protein domains are present. Ligand binds to these extracellular regions. Each RTK
sub-family binds a defined set of ligands, defined by the receptor extracellular regions. A
single-pass, α-helical transmembrane domain connects the extracellular regions to a short
juxtamembrane region, followed by a tyrosine kinase domain. A variable-length, unstructured
C-tail region, rich in substrate tyrosines, lies at the C-terminus of the receptor protein.
Biological roles
Because the diversity of the RTK family resides primarily in the extracellular ligand-sensing
machinery, RTKs are divided into sub-families based on these extracellular regions(Lemmon &
Schlessinger, 2010). Within each human RTK sub-family, there may be as few as one family
member, or as many as fourteen. In total, humans express 58 different RTKs, divided into 20
sub-families. Recent bioinformatic analyses have suggested an even broader family of RTKs is
expressed in unicellular organisms, but the relevance of these to metazoan RTKs is unclear
4
sub-family correlates roughly with the complexity of the body plan and organism size. For
example, C. elegans and D. melanogaster express only one ErbB sub-family receptor, whereas
mammals express four(Stein & Staros, 2006). Similarly, humans express four fibroblast growth
factor (FGF) receptors and 22 different FGF ligand molecules, where C. elegans expresses just
two FGF ligands and one FGF receptor(Itoh, 2007). In light of this observation, it is not surprising
that RTKs seem to function primarily as master regulators of tissue development.
Each RTK sub-family is generally associated with the development of a particular subset
of tissues or cell types. Diversity within RTK sub-families adds another layer of regulation and
complexity. In the well-studied insulin receptor family of RTKs, genetic ablation of the signaling
pathway universally results in growth retardation, although the magnitude and mechanism of the
effect depends on the specific receptor or ligand targeted. In mammals, knockout of the IGF-1
growth factor (which stimulates the IGF-1 receptor) results in decreased bone cell proliferation
and a severe growth retardation, whereas insulin receptor (IR) knockout results in a mild growth
retardation, due to a slight decrease in adipose tissue mass – with no effect on early bone
development(Nakae et al, 2001). Similarly, within the mammalian PDGF receptor family, receptor
deletion results in severe developmental defects and embryonic or early post-natal death due to
insufficient numbers of specific smooth muscle cell populations in the vasculature. Mice null for
PDGF-A (a PDGFRα ligand) lack a population of smooth muscle cells that normally participates
in formation of lung alveoli, and these mice display an emphysema-like phenotype at birth. For
PDGF-B (a PDGFRβ ligand), knockout results in embryonic death due to massive
hemorrhaging(Betsholtz, 2004). Hemorrhaging occurs because PDGF-B is required for the
expansion of a vascular smooth muscle cell population that forms the primary wall structure of
blood vessels. In yet another illustration of this theme, Trk receptor family members are required
for development of different neural tissues; TrkA for nociceptive sensory neurons (TrkA-null mice
are insensitive to pain), and TrkB for the vestibular ganglion (TrkB-null mice exhibit defects in
5
cellular differentiation and proliferation events underlying development, following a prototypical
axis in which ligand and receptor act as an autonomous, modular signaling unit.
In contrast to the well-defined and autonomous pathways described above, it must be noted that
several RTKs require other accessory proteins, co-receptors, or higher-order oligomerization
(beyond dimerization) in order to transmit their signals. For example, the highly-conserved kinase
activity within the intracellular domain of RTKs is severely compromised or absent in 8 out of the
58 human RTKs: Ryk, Ror1, Ror2, ErbB3, CCK4, EphA10, EphB6, and SuRTK106(Mendrola et
al, 2013; Shi et al, 2010). For ErbB3 and EphB6 in particular, there is good evidence that these
receptors can interact with other autonomous RTKs to modulate signaling, perhaps by activating
them allosterically (Citri, 2003; Truitt & Freywald, 2011). Furthermore, accumulating evidence
suggests that several RTKs function as Wnt receptors or co-receptors. To date, the RTKs MuSK,
CCK4, Ryk, and Ror2 have been implicated in Wnt signaling(Niehrs, 2012). Finally, higher-order
oligomerization is known to modulate signaling by the Eph, DDR, and Tie receptors(Lemmon &
Schlessinger, 2010; Mihai et al, 2009). Understanding RTK signaling through non-autonomous
pathways remains a highly active field of research, and these alternative signaling modes remain
poorly understood.
Roles in cancer and other diseases
Given the widespread role of RTKs in driving proliferative processes in development, it is no
surprise that anomalous regulation of RTKs underlies a wide variety of disease states. Where
loss-of-function mutations underlie diverse developmental and degenerative pathologies,
gain-of-function mutations are largely associated with cancer development and progression (owing to
unchecked cell proliferation and survival). Indeed, the ErbB and Met RTK sub-families (among
others) were initially discovered as homologs of viral oncogenes(Dean et al, 1985; Sergeant et al,
1982). The important role of RTK signaling in cancer is reflected by the widespread development
and clinical use of RTK inhibitors for cancer therapy. To date, of the 20 RTK sub-families, only 5
6
The majority of these ‘non-targets’ are kinase-compromised (Ror, Ryk, CCK4, and SuRTK106),
and the remainder (MuSK and LMR) are not known to signal through autonomous ligand-receptor
complexes(Mendrola et al, 2013). Among the 14 RTK sub-families targeted in cancer therapy, 11
are targeted by clinically-approved inhibitors, while the remaining three families (Trk, Axl, and
Insulin receptor family) are targets of active clinical development(Alamgeer et al, 2013;
D'Arcangelo et al, 2013; Festino et al, 2013; Haisa, 2013; Heldin, 2013; Hynes, 2009; Montero et
al, 2011; Norris et al, 2011; Paccez et al, 2014; Scagliotti et al, 2013).
Two major challenges for therapeutic RTK inhibition are well-known: specific targeting of RTK
kinase activity is difficult to achieve due to the relative conservation across the human
kinome(Festino et al, 2013; Gao et al, 2013), and resistance to RTK-directed therapies typically
develops on a timescale of weeks or months(Hynes, 2009; Rosenzweig, 2012). Most
RTK-directed small molecule therapeutics inhibit multiple kinases. A broad range of kinase inhibition
may actually be useful in many disease contexts, but it severely complicates the drug
development process. For example, off-target kinase inhibition may underlie undesirable side
effects that limit dosing below a crucial therapeutic threshold. RTK-directed therapeutic
antibodies sidestep the kinase specificity problem by targeting the diverse extracellular regions of
receptors with exquisite specificity. Unfortunately, however, both therapeutic antibodies and
small molecules directed at RTKs rapidly lose efficacy in treated patients. The acquisition of
specific, recurrent resistance mutations is a rapidly developing paradigm in cancer
therapy(Rosenzweig, 2012). One promising approach to combat resistance is rational
combination therapy, in which drugs targeting a likely resistance pathway are co-administered
with a drug targeting the primary oncogenic driver(Kwong & Davies, 2014). Additionally,
expanding our mechanistic understanding of RTK signaling should allow us to more potently and
specifically target aberrant RTK signaling in cancer and other diseases.
Whereas cancer therapeutics are designed to inhibit diverse RTK signaling pathways, there is a
7
contexts. The classic example, of course, is insulin therapy for diabetes. Beyond insulin, Tie1
receptor stimulation has been investigated as a mechanism of supporting vascular integrity in
sepsis(David et al, 2013). Therapeutic potential for HGF, the ligand for Met, has been revealed in
both a topical form for recurrent leg ulcers(Nayeri et al, 2002), as well as in gene therapy for
treatment of chronic limb ischemia(Shigematsu et al, 2010). Indeed, considering the widespread
role of RTKs in vascular development, several RTK ligands have been investigated as therapies
for ischemia(Hammer & Steiner, 2013). In addition, great excitement has surrounded the recent
discovery that Nrg1, an ErbB ligand, stimulates cardiomyocyte generation in the adult heart and
aids recovery from heart failure(Bersell, 2009; Galindo et al, 2013; Hao et al, 2014). The clear
therapeutic promise of receptor tyrosine kinase drugs – whether inhibitors or agonists, specific or
broad, small molecule or antibody – has prompted detailed mechanistic studies of RTK signaling.
General mechanisms of signaling by receptor tyrosine kinases
At the most general level, RTKs act as transmembrane receptors that convey an extracellular
signal into the interior of the cell. RTK extracellular regions (herein referred to as ECRs) directly
bind to a ligand, resulting in increased effective kinase activity within the intracellular tyrosine
kinase domain of the receptor (Fig. 2). This is the core process of RTK signaling. The increased
effective kinase activity of RTKs contributes to intermolecular receptor trans-autophosphorylation,
in which one receptor molecule phosphorylates another at defined tyrosine residues.
Phosphotyrosines on the receptor are then specifically recognized by interaction modules in other
signaling molecules that assemble active signaling complexes upon binding to activated receptor.
These modules include Src homology 2 (or SH2) domains and phosphotyrosine binding (or PTB)
domains. Activated RTK signaling complexes exploit a handful of well-characterized pathways to
broadly modulate gene expression, directly giving rise to the multitude of proliferative or
differentiation events triggered by RTKs. All the while, physiological RTK signaling is regulated at
many levels. Expression of both receptor and ligand is tightly controlled. Constitutively active
8
receptors (Sastry & Elferink, 2011). Upon activation of receptor, down-regulation occurs through
of variety of mechanisms.
Figure 2: RTK ligands induce dimerization by diverse structural mechanisms. Upon dimerization,
the kinase domains within a dimer become activated. An activated receptor kinase within one
protomer proceeds to phosphorylate substrate tyrosines on its dimeric partner. Receptor
phosphorylation generally increases the inherent kinase activity (although ErbBs are an exception
to this rule). Receptor phosphotyrosines serve as docking sites for PTB- and SH2-domain
containing proteins, which seed active signaling complexes.
Ligand-binding and dimerization
For the vast majority of autonomous RTKs, ligand-binding and receptor dimerization are
intimately-linked processes that are thought to be tightly coupled energetically. The single
well-established exception to this rule is the insulin receptor family, in which receptors are
9
contexts, the ErbB family is prone to form ligand-independent dimers as well; this idea will be
further examined later, and indeed is a primary focus of this thesis and the active work of others
in the field (Chung, 2010; Gadella & Jovin, 1995; Saffarian, 2007; Tao & Maruyama, 2008; Webb,
2008; Yu, 2002). Incidentally, both of these receptor families are now known to utilize a unique
structural mode of dimerization, in which the receptor dimer interface is formed exclusively by
receptor protein surfaces; the contribution of ligand to dimerization is indirect(Garrett, 2002; Liu et
al, 2012a; Ogiso, 2002). At the opposite end of the spectrum, the ligand-dependent, extracellular
dimerization interface that activates Trk receptors is exclusively ligand-mediated, with ligand
acting as a bivalent, non-covalent cross-linker for two receptor molecules (Banfield et al, 2001;
Wiesmann et al, 1999).
Between these two extremes lie all the other structurally-characterized RTKs. For these
receptors, the homo-dimeric contacts that stabilize formation of an active dimer are distributed
between both receptor-receptor contacts and ligand-ligand contacts, and even ligand-accessory
molecule contacts in the case of FGFR, where the FGF ligands bind receptor and heparin
concurrently(Ibrahimi et al, 2005). The receptors Kit and VEGFR are two prototypical examples
in which ligand-ligand contacts (within a bivalent ligand dimer) as well as receptor-receptor
contacts contribute to the stability of active receptor dimers(Brozzo et al, 2012; Leppanen et al,
2013; Yuzawa et al, 2007). The relative topology of ligand-ligand and receptor-receptor contacts
in these receptors (as well as PDGFR) is remarkably well-conserved(Yang et al, 2008). The
receptor-receptor contacts typically involve membrane-proximal regions of the receptor, whereas
the ligand-ligand contacts involve more distal domains of the receptor, away from the membrane.
There is some evidence that this sort of dual dimer-interface topology is important in ErbB
receptors as well, even though both of the interfaces in this case are completely
receptor-mediated(Liu et al, 2012a; Lu et al, 2010; Moriki et al, 2001). Although the relevance of this
topology is not completely understood, there is some evidence that the membrane-proximal
10
relationship, rather than simply increasing dimerization affinity(Brozzo et al, 2012; Yang et al,
2008).
Kinase domain activation and trans-autophosphorylation
Just as the ECRs of RTKs are activated through dimerization, so too are the intracellular kinase
domains (Fig. 2). Diverse mechanisms underlie the dimerization-dependent kinase activity of
RTKs, and there is no general correlation between the respective modes of ECR dimerization and
kinase activation within the RTK family. Broadly speaking, kinase domains adopt an array of
differently autoinhibited inactive conformations. Receptor dimerization converts these inactive
states, by a variety of different mechanisms, into an active kinase conformation that is largely
conserved throughout the RTKs(Huse & Kuriyan, 2002). For the ErbB receptor EGFR,
dimerization of the kinase domain occurs in an asymmetric ‘head-to-tail’ arrangement that
allosterically activates the kinase domain of one protomer(Zhang, 2006).
For all the other well-characterized RTKs, auto-inhibitory intramolecular interactions suppress
kinase activity by sterically blocking the active site; for these kinases, activation requires
phosphorylation of key residues to interrupt the auto-inhibitory interaction(Lemmon &
Schlessinger, 2010). The residues involved in auto-inhibition may lie in the ‘activation-loop’
sequence within the kinase domain, the membrane-proximal juxtamembrane region (N-terminal
to the kinase domain), or within the disordered tail of the protein, C-terminal to the kinase domain.
For these kinases, it’s thought that a basal level of kinase activity (even when auto-inhibited) is
sufficient to explain the phosphorylation events responsible for reversing auto-inhibition. Within
the high local concentration of receptor afforded by dimerization, this low level of activity is
sufficient to trigger receptor activation through a first initial trans-autophosphorylation step.
Following this first trans-autophosphorylation event within a dimer, an ordered and efficient
11
The number and location of trans-autophosphorylation sites varies by receptor. Most are located
in the C-terminal tail, the juxtramembrane region, or in the so-called kinase insert domain. Some
of these sites serve as direct docking sites for the SH2 and PTB domains of signaling molecules
including PI(3)K, Shc, and Grb2(Schulze et al, 2005). Other sites may serve as docking sites for
receptor-specific ‘substrate’ proteins that function as scaffolding proteins; for these receptors,
these substrate proteins are required for the full complement of signal transduction. Such
substrate proteins include the insulin receptor substrate (IRS) proteins, and the FGF receptor
substrate (FRS) proteins. There is emerging evidence that ligand identity and co-receptor
engagement may modify the receptor phosphorylation pattern in a way that directs or modulates
aspects of downstream signaling(Hartman et al, 2013; Wilson et al, 2012a). Inevitably, RTKs
directly stimulate signaling via the ERK and AKT pathways to regulate gene expression patterns.
RTK modulation of STAT transcription factors has also been reported, but the mechanistic
onnection is less clear and may be indirect(Gao et al, 2007).
Exogenous regulation of RTK signaling activity
A host of accessory factors in the cell serves both to suppress basal RTK signaling activity in the
absence of ligand stimulation, and to down-regulate or terminate RTK signaling activity on a
timescale of minutes. Two well-known systems largely fulfill these roles. One is the protein
tyrosine phosphatase family (PTPs), which exhibit constitutive activity at the cell membrane to
convert spurious phosphotyrosines back to benign tyrosine residues and to reverse the effects of
ligand activation (Sastry & Elferink, 2011). In addition, activated receptors are internalized by
endocytic pathways, and are either degraded in the lysosome or de-activated by exposure to low
pH in early endosomes before de-phosphorylation by PTPs and subsequent recycling to the
plasma membrane.
The PTP1B protein is a well-established suppressor of basal signaling for EGFR, Met, PDGFR,
and IGF1R(Sastry & Elferink, 2011). It has long been appreciated that general PTP inhibition (for
12
targeted studies suggest that PTP1B is primarily responsible for this effect (Elchebly et al, 1999;
Tran et al, 2003). PTPs in general, typified by PTP1B, seem to exhibit constitutive activity with
very broad specificity(Stuible & Tremblay, 2010). Given this apparent lack of PTP regulation, it
should not be surprising that PTPs play a very complex and convoluted role in vivo. In fact, it is
clear that PTP1B is required for transforming activity in some contexts, so it is not simply a global
inhibitor of signaling, but may have signal-promoting properties as well(Bentires-Alj & Neel,
2007). Another PTP, known as SHP2 or PTPN11, possesses two phosphotyrosine-binding SH2
domains, which confer the ability to bind directly to RTK signaling complexes via direct interaction
with the Gab adaptor proteins; however, SHP2 is not known to play any inhibitory role in RTK
signaling, and instead seems to promote a variety of downstream signaling pathways(Sastry &
Elferink, 2011). Finally, the PTPN12 protein has also been shown to down-regulate RTK
signaling pathways via its phosphatase activity, but it is not known whether it acts primarily on
downstream signaling effectors or directly at the RTK(Charest et al, 1997).
Once an RTK has been activated by ligand-induced trans-autophosphorylation, the receptor must
be inactivated on an appropriate timescale to avoid perpetual signaling. This inactivation is
initiated primarily by the endocytosis. Activated receptors are shuttled to early endosomes. In
some cases this results in ligand dissociation (due to low pH) and subsequent recycling of
de-activated receptor to the plasma membrane. In other cases, low pH does not cause ligand
dissocation, and the receptor is sorted to lysosomes where the receptor is proteolyzed and
degraded. Many different mechanisms have been implicated in activity-dependent RTK
endocytosis, but the primary route under normal physiological conditions is though to depend
largely on clathrin-mediated endocytosis (CME)(Goh & Sorkin, 2013). Every major RTK family is
known to become ubiquitinated, but the role of direct ubiquitination in CME is receptor-dependent.
For example, whereas EGFR and FGFR-2 endocytosis do not depend on receptor ubiquitination,
IGF1R endocytosis requires it(Mao et al, 2011). When the concentration of activated receptor at
the cell surface reaches extremely high levels (as in the case of cancer cells overexpressing
13
pathways become relevant, although little is known about the relevance of these pathways in
normal physiological conditions (Wiley, 1988).
Biology of ErbB family RTKs
Receptor knockout studies elucidate roles in cardiac and neural development
Individual genetic deletion of the four mammalian ErbB receptors in mice demonstrates that all of
these receptors play important roles in normal cardiac and neural development. Global knockout
of ErbB2 or ErbB4 results in embryonic lethality at day E10.5 due to defective cardiac trabeculae
formation, a process that depends on myocyte proliferation. ErbB4 deletion also compromises
development of hindbrain tissue, whereas ErbB2 deletion affects development of cranial neural
crest-derived sensory ganglia(Gassmann, 1995; Lee, 1995). Knockout of ErbB3 causes
embryonic lethality at day E13.5, with pups exhibiting blood reflux through defective valves due to
aberrant cardiac cushion formation. Trabeculae formation appeared normal for the ErbB3-null
mice, but the hindbrain was once again severely compromised(Erickson, 1997). Compared to the
other ErbB receptors, ErbB1/EGFR seems to play a more global role in development. EGFR
deletion has strain- or background-dependent effects, resulting in either embryonic death due to
placental abruption, or death three weeks after birth due to multiple organ failure, with defects in
the skin, kidney, brain, liver, and the gastrointestinal tract(Threadgill et al, 1995). Rescue of the
strain-specific placental defect showed that a broad, strain-independent neurodegeneration
program occurs soon after birth(Sibilia et al, 1998). EGFR knockout mice also exhibit enlarged
cardiac valves(Chen, 2000).
Ligand knockout studies display complex phenotypes
Not surprisingly, genetic deletion of ErbB ligands phenocopies ErbB receptor deletion in certain
contexts; however, it is equally clear that some ligands are redundant in certain contexts. There
are nine discrete genes encoding a total of 11 distinct ErbB ligands (each Nrg gene has 2 splice
14
mice, the most striking phenotypes belong to HB-EGF- and Nrg1-null mice. Global knockout of
HB-EGF, a ligand for EGFR and ErbB4 that is expressed in cardiomyocytes, causes severe
cardiac dysfunction typified by malformed heart valves (analogous to EGFR deletion) and
considerable enlargement of the heart(Iwamoto, 2003; Jackson et al, 2003). Although
administration of HB-EGF to these mice caused increased phosphorylation of ErbB2 and ErbB4
as well as EGFR, phenotypic comparisons indicated that Nrg1 is most likely to be the crucial
ligand for ErbB2-heterodimer signaling in the heart. Genetic deletion of Nrg1 produces mice with
defects in formation of trabeculae (as in ErbB2- or ErbB4-null mice) and the cardiac cushion (as
in ErbB3-null mice)(Meyer, 1995). Additionally, Nrg1-null mice exhibit defects in cranial ganglia
and Schwann cell development, revealing an essential role for Nrg1 in ErbB2, ErbB3, and ErbB4
signaling in the developing brain. Although the low-affinity Nrg1α isoforms constitute the bulk of
Nrg1 mRNAs in cardiac endothelial cells, specific knockout of these isoforms does not appear to
impair heart development, suggesting that the high-affinity Nrg1β isoforms are primarily
responsible for ErbB2-, ErbB3-, and ErbB4-mediated developmental cues in the heart(Cote,
2005; Li, 2002). In addition to their role in cardiac development, ErbB2, ErbB4, and Nrg1 are
crucial for adult cardiac function. Dilated cardiomyopathy occurs in mice with a cardiac-specific
conditional knockout of ErbB2(Crone, 2002; Ozcelik, 2002) or ErbB4(Garcia-Rivello, 2005); both
of these receptors are expressed in adult cardiomyocytes. Cell-based experiments lend further
credence to the notion that Nrg1 signaling through ErbB2/ErbB4 complexes plays a unique role in
cardiac physiology. Moreover, cardiac toxicity has emerged as one of the most problematic side
effects of ErbB2-targeted cancer therapy (Guglin et al, 2008).
Phenotypes for other ErbB ligand-knockout mice are more convoluted. In contrast to the
dramatic cardiac effects of Nrg1 deletion, Nrg2 knockout mice are viable, and exhibit growth
retardation and inefficient reproduction(Britto et al, 2004). TGFα deletion yields a ‘wavy-hair’
phenotype, accompanied by abnormal follicle morphology(Luetteke et al, 1999). Individual
deletion of amphiregulin (Arg) results in defects in mammary gland development; deletion of EGF
15
exacerbated mammary defect(Luetteke et al, 1999). Even mice triple-null for EGF, Arg , and
TGFα survive to maturity. The final three ErbB ligands, Betacellulin (Btc), Epigen (Epg), and
Epiregulin (Erg), have all been deleted in mice, and none of the resulting knockout mice display
overt defects in development(Dahlhoff et al, 2013; Jackson et al, 2003; Lee et al, 2004). The
striking contrast in phenotypes for receptor deletion compared to ligand deletion (especially for
EGFR, compared to its seven cognate ligands) has sparked the view that most of the ErbB ligand
functions are covered by several-fold redundancies and constitute a robust developmental
signaling sysem.
Beyond development, recent studies have pushed forward our understanding of ErbB functions in
mature animals. These studies have primarily agreed with the view that most ligands are
redundant. For example, Epg, Erg, and Betacellulin have all been show to stimulate oocyte
maturation in vivo(Park et al, 2004). In response to corneal wounds, the ligands TGFα, Arg, and
Betacellulin are all upregulated, and each can contribute individually to the wound healing
response(Zieske et al, 2000). Another striking hint at the combinatorial nature of ligand function
comes from genomic conservation: the genes encoding Epg, Erg, Arg, and Betacellulin are all
located adjacent to each other in mammal genomes(Lee et al, 2004). This pattern in consistent
both with a recent origin for these ligands via gene duplication, and with common regulation at the
chromatin level.
Mechanistic insight into ErbB signaling
Signaling by the ErbB family underlies developmental processes in metazoans, from D.
melanogaster and C. elegans to mammals(Freeman, 1997; Hill, 1992). In adults, ErbB signaling
plays a crucial role in homeostasis of the cardiac and reproductive systems as well as wound
healing; aberrant ErbB signaling contributes to the development of cancer, heart disease, and
neurological disorders. Indeed, genes encoding EGFR and ErbB2 were first identified as
oncogenes in the 1980s and have been viewed as important therapeutic targets in cancer ever
16
phosphotyrosine-based signal transduction in the 1980s, has motivated detailed mechanistic
characterization of ErbB receptor-mediated signaling. The ErbB/EGFR/HER family comprises
four receptors and eleven bona fide ligands in humans. ErbB receptors have a conserved
domain architecture, with the extracellular, ligand-binding region encoding alternating
-helix/solenoid (domains I and III) and cysteine-rich domains (domains II and IV in humans). The
intracellular region consists of a short juxtamembrane region, a tyrosine kinase domain, and a
regulatory but largely unstructured C-terminal tail. A single transmembrane helix connects the
extracellular region (or ECR) to the intracellular region (or ICR).
Ligand stabilizes active receptor dimers and oligomers
Groundbreaking studies employing cross-linking techniques with antibody-purified EGFR led to
the proposal that EGFR (and, by extension, other ErbBs and RTKs in general) are activated by
ligand-induced dimerization(Yarden & Schlessinger, 1987a; Yarden & Schlessinger, 1987b).
Although a complete mechanistic understanding of ligand-mediated receptor activation is lacking,
high-resolution crystallographic structural models of receptor fragments have greatly informed the
current view (Fig. 3). Crystal structures of all four of the ErbB receptor extracellular regions have
been solved in the absence of ligand(Bouyain, 2005; Cho, 2002; Cho, 2003; Ferguson et al,
2003; Garrett, 2003). The EGFR ECR structure has also been determined in the fully
ligand-occupied state, with either EGF or TGFα as the activating ligand (Garrett, 2002; Lu et al, 2010;
Ogiso, 2002), and a Nrg-activated ErbB4 ECR structure has recently been reported as well(Liu et
al, 2012b). All of the ligand-regulated receptors (EGFR, ErbB3, and ErbB4) exhibit a compact,
‘tethered’ conformation in the absence of ligand. The tethered conformation is stabilized by an
intramolecular interaction involving a protruding -hairpin on domain II and a small surface on
domain IV (adjacent to another, smaller, -hairpin). The tethering interaction comprises 4-5
hydrogen bonds involving conserved residues in domains II and IV. In ErbB2, which exhibits an
extended, untethered conformation in crystals and in solution(Dawson et al, 2007), these
17
the autoinhibitory tether, and may therefore remain constantly poised to heterodimerize with other
ligand-bound ErbB receptors.
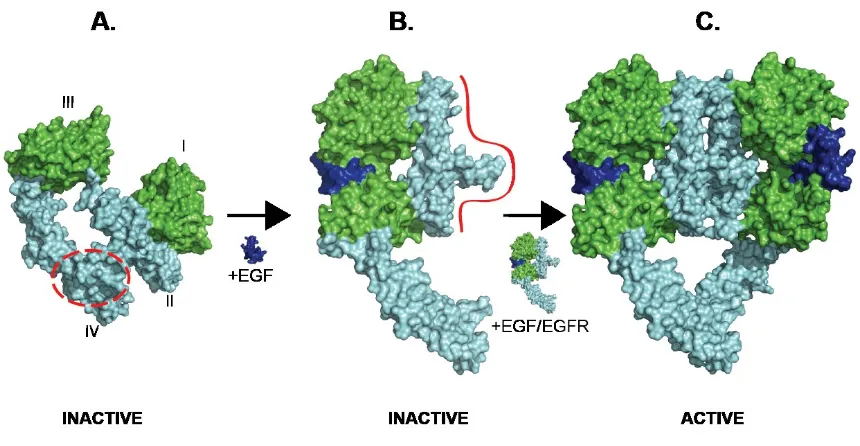
Figure 3: A model of ligand-induced dimerization and receptor activation based on crystal
structures and solution scattering data. A: tethered EGFR (adapted from crystal structure, PDB
1NQL) B: ligated, extended, monomeric EGFR, inferred from low-resolution SAXS data (Dawson
et al, 2007) C: ligated dimeric EGFR (adapted from crystal structure, PDB 3NJP).
The independent structures of ligand-bound (presumably activated) EGFR and ErbB4 ECRs
show largely conserved ligand:receptor and receptor:receptor interactions(Garrett, 2002; Liu et al,
2012b; Ogiso, 2002). Ligand binds to surfaces fromthe -helix/solenoid domains I and III, and
stabilizes a rearrangement of the ECR from a tethered to an ‘extended’ conformation. The ligand
fold is stabilized by three characteristic intramolecular disulfide bonds, though much of its
receptor-interaction surface is contributed by an otherwise unstructured C-terminal extension.
The extended, ligand-bound receptor is competent for dimerization as illustrated in Fig. 3. It is
striking that the dimerization interface is dominated by the -hairpin ‘tether’ from domain II, which
also stabilizes the inactive conformation. Indeed, mutating the domain II -hairpin compromises
18
dimerization interface is made by residues near the N-terminal part of domain II. Importantly, the
EGF- and TGFα-bound EGFR structures differ significantly in this N-terminal part of the domain II
dimer interface. The origin and significance of this structural difference is currently unclear; it
may arise simply from crystal packing differences, or as a consequence of the different receptor
protein boundaries used in the two structures. Alternatively, and as suggested by recent a recent
study employing a conformation-specific imaging technique in cells (Scheck et al, 2012), the
distinct ligands may inherently specify slightly different dimeric interfaces. This could explain
reported ligand-specfic signaling responses (Wilson et al, 2012a). Biophysical studies argue that
this portion of the dimer interface makes a small contribution to the overall dimerization
energy(Dawson, 2005). However, this does not mean that the structural difference is not
biologically relevant. Indeed, membrane-proximal contributions to dimerization of the Kit receptor
have been shown to make little or no contribution to dimerization affinity per se (Lemmon et al,
1997), but they do make crucial contributions to Kit activation – both by ligand and by oncogenic
mutations (Yuzawa et al, 2007).
ECR structures from the ErbB family argue that dimerization underlies ErbB signal transduction
(Fig. 3). Structures of intracellular regions argue that formation of an asymmetric dimer of the
tyrosine kinase domains, in which one kinase domain allosterically activates the other, allows
signal transduction to occur(Zhang, 2006). The juxtamembrane region of the receptor also
contributes to the stability of this asymmetric dimer(Red Brewer et al, 2009). However, it is
impossible at this time to fully reconcile our understanding of the ligand-mediated extracellular
signaling events with the details and intricacies of intracellular kinase domain regulation (Lu et al,
2012; Mi et al, 2011; Moriki et al, 2001; Roberts et al, 2012). Domain IV of the ECR is highly
flexible, especially in activated receptor dimers, so the orientation of receptors relative to the
membrane remains an open question. It has also been reported, using detergent-solubilized
receptors, that the ligand-bound ECR can form an active dimer structure while the inhibitor-bound
19
While the structural models described above can inform and explain ligand-induced dimerization,
kinase domain activation, and subsequent signal transduction, a wealth of observations
suggesting that current structural models are far from sufficient. First, numerous experimental
methods have indicated the existence of pre-formed, inactive receptor dimers in the absence of
ligand(Gadella & Jovin, 1995; Saffarian, 2007; Webb, 2008; Yu, 2002). This observation
undermines the simple model of receptor activation through ligand-induced dimerization. The
notion of physiologically-relevant, inactive, ligand-independent receptor dimers is bolstered by
recent crystal structures and biochemical studies of the D. melanogaster EGF receptor(Alvarado
et al, 2010; Alvarado, 2009). Second, the complex binding characteristics of EGF and other ErbB
ligands to cell-surface receptors, first noted 30 years ago, can not be explained based on the
available structures(MacDonald, 2008; Ozcan, 2006; Shoyab, 1979). It now appears that the
complex ligand-binding characteristics reflect negative cooperativity, as seen in many receptor
families (De Meyts, 2008). The origin of this cooperativity – and its modulation by intracellular
modifications (MacDonald-Obermann, 2009) – cannot be explained by simple dimerization
models, because the crystal structures of human EGFR display symmetric ligand-binding sites.
Third, there is evidence that distinct ErbB ligands induce distinct receptor responses at the level
of phospho-tyrosine site usage and endocytosis kinetics(Hobbs, 2002; Roepstorff et al, 2009;
Wilson et al, 2012a); these data suggest ErbB receptors can discriminate between ligands, which
implies that multiple different activated receptor states may coexist.
In combination, these three observations (ligand-independent dimers, negatively-cooperative
ligand-binding, and ligand-specific receptor responses) strongly challenge the prevailing
structure-based signaling model in which ligand-induced dimerization is the essence of ErbB
receptor activation. These challenges argue that subtle, unappreciated modes of allosteric must
contribute importantly to the regulation of ErbB receptor activation. The goal of my thesis was to
further elucidate how ligand activates the EGF receptor through unappreciated allostery.
20
families, and it could also further inform the design of more effective ErbB-targeted therapeutics
21
22
Pre-dimerized EGFR requires ligand to form a domain II-mediated dimer
The structural model depicted in Figure 3 implies that EGFR activation is reducible to a
ligand-induced dimerization event. In contrast, recent findings from the D. melanogaster system
imply that EGFR activation requires induced allosteric changes within a pre-formed,
ligand-independent dimer(Alvarado et al, 2010). Extensive support for each of these models has been
reported. These two distinct models for understanding how ligand activates EGFR are not
mutually exclusive, but they do have very important implications for therapeutic targeting of
EGFR. For example, development of therapeutic antibodies against EGFR has, to date, focused
on selecting antibodies that prevent receptor dimerization. If stable, inactive EGFR dimers form
in tumors, then antibodies that stabilize such a complex might provide a unique therapeutic effect.
While there is evidence that inactive, ligand-independent EGFR dimers are ubiquitous in a
cellular context, the isolated human EGFR ECR (utilized for biochemical, biophysical and
structural studies) does not form a ligand-independent dimer. To better understand how ligands
can activate ligand-independent (‘pre-formed’ or ‘pre-dimerized’) EGFR dimers, we genetically
fused a dimerizing Fc domain to the EGFR ECR for in vitro analyses. This pre-dimerized EGFR,
termed sEGFR-Fc, was amenable to thermodynamic, biophysical, and structural studies.
To determine the gross domain arrangement of pre-formed EGFR dimers, we collected
and analyzed electron microscopy (EM) images utilizing uranyl formate negative-stain to generate
contrast. Class-averaging of single particles obtained from images of the sEGFR-Fc protein in
complex with EGF yielded clear densities for all 4 receptor domains as well as the Fc domain and
EGF (Fig. 4A). The ligand-receptor complex itself bears a striking resemblance to previously
published EM class-averaging results, for both isolated receptor ECRs and for near full-length
receptors reconstituted in detergent micelles(Mi et al, 2011). In a subset of classes, the
Fc-domain appears well-resolved with good signal, indicating that the particular arrangement of the
Fc domain relative to the receptor domains represents a highly-populated state. The relative
23
domains in near full-length receptors. Thus, it is clear that pre-formed human EGFR dimers can
25
Figure 4: A: Reference-free class averages generated in the Spider software suite, using
single-particle EM images of EGF:sEGFR-Fc complexes. B: Rg values derived by Guinier analysis of
SAXS data, or calculate from the structural models described in D-F using the HydroPro software.
SAXS scattering was recorded for 10-20 µM sEGFR-Fc in the presence or absence of a 1.3-fold
excess of EGF. C: Dmax determinations, as previously described. For B.) and C.), four
independent experiments were performed, and the mean is plotted with error bars representing
the standard error of the mean (SEM). Paired t-tests were used to compare SAXS parameters in
the presence or absence of EGF, and the p-values are indicated. D-F: Distinct structural models
were constructed; Rg and Dmax values were back-calculated from these models using HydroPro,
for comparison to experimentally-determined parameters for sEGFR-Fc.
sEGFR-Fc without added ligand was also imaged by electron microscopy and particles were
subjected to the same class-averaging algorithms as used for ligand-bound sEGFR-Fc (Fig. 5); in
contrast to the results for the ligand-bound complex, sEGFR-Fc alone failed to generate
signal-enhanced class averages with well-defined and interpretable inter-domain relationships (even
when using the exact same protein preparations). This result reveals that a well-ordered,
back-to-back dimer, mediated by interactions involving domain II of each protomer, does not form in
the absence of ligand. It is not clear whether the individual receptor molecules with the
Fc-mediated dimer maintain a tethered or extended conformation, or represent an ensemble that
samples multiple conformations. These data do argue, however, that forcing dimerization of the
EGFR extracellular region (by Fc fusion) does not simply drive it to form domain II-mediated
dimers of the sort seen in our crystallographic studies of the D. melanogaster EGFR (Alvarado et
26
Figure 5: An example of a raw EM image file (in this case, sEGFR-Fc with no ligand) used for
single-particle analysis. For more details, see experimental methods.
In an effort to better understand the allosteric effect of ligand-binding on pre-formed
human sEGFR dimers, we also performed SAXS experiments on sEGFR-Fc in the presence and
absence of EGF. For sEGFR-Fc in complex with EGF, we determined the radius of gyration (Rg)
27
previously(Dawson et al, 2007). In the presence of EGF, the experimentally-determined
scattering parameters are in good agreement with the parameters predicted from a structural
model in which the previously observed back-to-back extended receptor dimer has an Fc domain
dimer appended at the C-terminus of EGFR domain IV, with the Fc dimer extended away from
the receptor (Fig. 4B-D). In contrast, the sEGFR-Fc protein alone generates a scattering curve
with a significantly larger Rg and Dmax. These scattering parameters are most consistent with
structural models in which the two receptor protomers in the unligated sEGFR-Fc dimer are not
interacting in an ordered back-to-back dimer (Fig. 4E,F). In agreement with EM data, these
SAXS data suggests that sEGFR-Fc – and, by extension, physiological EGFR pre-formed dimers
– requires ligand binding in order to form the domain II-mediated dimerization interface seen
crystallographically for human and Drosophila EGFR. This conclusion is further supported by
recently published EM data on near full-length ligand-independent EGFR dimers – in which
dimerization was driven by an EGFR kinase inhibitor rather than an Fc-fusion approach (Lu et al,
2012). In that case, antibody-labeling showed that the extracellular regions of the protein were
conformationally heterogeneous. The notion that ligand-independent EGFR dimers require ligand
to form a stable, domain II-mediated dimer stands in surprising contrast to the allosteric activation
mechanism describes for D. melanogaster EGF receptor (Alvarado et al, 2010). Given our
surprising findings, we sought to re-examine other key assumptions about
dimerization-dependent EGFR regulation, which were originally inferred from the structure-based model
presented in Figure 3.
EGFR dimerization does not stabilize ligand-binding
As outlined in Fig. 3, the EGFR ECR monomer adopts a compact, tethered structure in the
absence of ligand. The intramolecular tethering interaction sterically blocks receptor dimerization
by occluding the domain II dimerization interface. Upon ligand-binding, receptor domains I and II
move with respect to domains III and IV such that ligand can bind to surfaces on domains I and III
28
the domain II/III connection) exposes the domain II dimerization arm as shown in the middle
panel of Fig. 3, and two 1:1 receptor:ligand complexes then form a symmetric homodimer (Fig.
3C) with a dissociation constant of ~1 µM(Dawson, 2005). This corresponds to a Gibbs free
energy (∆G) of -7.5 kcal/mol for dimerization. Several crystal structures support the views
depicted in Figure 3 A and C for both EGFR and ErbB4(Bouyain, 2005; Burgess, 2003; Liu et al,
2012a). The structure of a ligand-bound, monomeric receptor has not been determined at atomic
resolution and physiologic pH, but small-angle x-ray scattering (SAXS) data argue strongly that
such a complex can adopt an extended, non-tethered conformation (as depicted in Fig. 3B),
grossly reminiscent of one half of a 2:2 ligand:EGFR dimer(Dawson et al, 2007).
To examine the energetic linkage of ligand-binding and dimerization for EGFR, we exploited
isothermal titration calorimetry (ITC) to compare the thermodynamics of ligand-binding to either
wild-type EGFR ECR (sEGFRWT, residues 1-618 of mature human EGFR) or the Y251A/R285S
variant (sEGFRY251A/R285S). sEGFRY251A/R285S does not dimerize upon ligand-binding, and this
combination of mutations also abolishes signaling by the full-length receptor (Dawson, 2005;
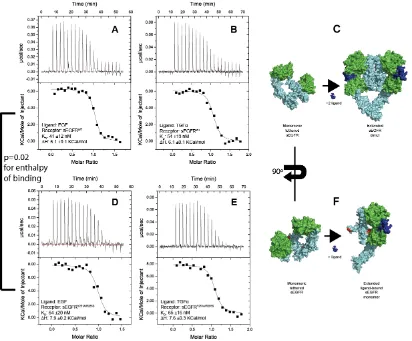
Ogiso, 2002). Titration of the ligand EGF into sEGFRWT (Fig. 6A) and into sEGFRDomainIII (Fig. 7A)
is consistent with previously published data. Without the benefit of dimerization-deficient variants,
theseearlier studies interpreted wild-type titrations to represent enthalpically-driven
ligand-binding and strongly entropically-driven receptor dimerization(Alvarenga et al, 2012; Lemmon,
1997). It is clear, however, from the titration of EGF into the non-dimerizing sEGFRY251A/R285S
variant that ligand-binding itself is driven by strongly favorable entropic terms (Fig. 6C). This
finding calls for a reinterpretation of previous thermodynamic studies of EGF/EGFR interactions.
Based on a calorimetric dissociation experiment (Fig. 7B) the enthalpy of dimerization of
ligand-bound EGFR is <<|2| kcal/mol. Because the dimerization enthalpy is insignificant in this context,
sEGFRWT as well as sEGFRY251A/R285S titrations can be accurately fit with a single-site binding
model, where the binding event represents ligand binding to receptor. This thermodynamic
29
dimerization-deficient receptor – is maintained for the EGFR ligand TGFα, as well as EGF (Fig. 6,
Table 1).
Table 1
EGFR variant Ligand KD
Ligand-induceddimerization
, µM
KD
Ligand-binding
, nM
∆H
Ligand-binding,
kcal/mol
Wild-type
EGF
~1
41 ±12
+6.1 ±0.1
TGFα
~1
54 ±13
+6.1 ±0.1
Y251A/R285S EGF
>200
64 ±20
+7.9 ±0.2
TGFα
>200
65 ±16
+7.6 ±0.3
Figure 6: ITC titrations A: EGF titrated into sEGFRWT(5 independent experiments performed; 1
representative titration is shown) B: TGFα titrated into sEGFRWT (2 independent experiments
30
during titration of ligand into sEGFRWT D: EGF titrated into sEGFRY251A/R285S (3 independent
experiments performed; 1 representative titration is shown) E: TGFα titrated into sEGFRY251A/R285S
(2 independent experiments performed; 1 representative titration is shown) F. During ligand
titration into sEGFRY251A/R285S, ligand binds to the receptor and the tether interaction is released,
resulting in a relatively extended receptor conformation, but the receptor does not dimerize.
Positions Y251 and R285 are highlighted in red. An unpaired student’s T-test comparing the ΔH
value for EGF binding to sEGFRWT and sEGFRY251A/R285S determined a p-value of 0.02, indicating
high confidence that ligand-binding to sEGFRWT and sEGFRY251A/R285S is qualitatively and
significantly different.
Further insight into the relationship between ligand-binding and receptor dimerization can be
gained by comparing the overall affinity of EGF for sEGFRWT and sEGFRY251A/R285S. If the model
in Fig. 3 is accurate, and monomeric, ligated receptor mimics one half of a 2:2 receptor dimer,
then the Gibbs free energies of ligand-binding (-9.7 kcal/mol, based on a KD for ligand-binding of
41nM) and receptor dimerization (-7.5 kcal/mol, based on a KD for dimerization of ~1M) should
be additive; in other words, receptor dimerization should strongly stabilize ligand-binding, just as
ligand-binding stabilizes dimerization in this simple model. In the most extreme case, the KD for
ligand binding to dimerizing receptor should therefore be nearly 6 orders of magnitude lower than
for non-dimerizing receptor, corresponding to 7.5 kcal/mol of free energy gained from
dimerization. Contrary to this expectation, we find that the ligand-binding affinities for sEGFRWT
and sEGFRY251A/R285S are equivalent within the sensitivity of the ITC experiment (Table 1). This
finding agrees with recent studies of ligand-binding to full-length receptor in cells,in which model
fitting of EGF-binding data collected for cells expressing a wide variety of different receptor levels
indicated negative-cooperativity, but a similar ligand-binding affinity for monomeric and dimeric
receptor (MacDonald, 2008). The notion that monomeric and dimeric receptors bind EGF with
equivalent affinities is further supported by comparing the EGF-binding affinity for sEGFRWT
determined by ITC with that determined by a fluorescence anisotropy (FA) assay, as illustrated in
31
dimerization of ligand-bound sEGFR, so this assay effectively reports on EGF binding to
monomeric sEGFRWT. Nonetheless, EGF binds sEGFRWT with an affinity of 78nM in FA assays,
arguing that its affinity for sEGFR is not affected significantly by dimerization per se. Affinities for
EGF binding to wild-type receptor in ITC (where the receptor dimerizes) and in fluorescence
anisotropy assays (where sEGFR does not dimerize) are essentially the same, and equivalent to
that measured by ITC for the sEGFRY251A/R285S variant, arguing that receptor dimerization does
not stabilize ligand-binding.
Figure 7: A: ITC titration of EGF into 10µM sEGFR domain III. Fitting to a single binding-site
model yielded a KDof 3.1 ±1.9 µM, and a ∆H of -3.5 ±1.1 kcal/mol. B: To measure the heat of
ligand-induced dimerization, 17.5 µM sEGFRWT in complex with a 1.3-fold excess of EGF was
injected into an ITC cell containing only buffer. 13 injections of 3 µL were performed, allowing
measurement of dissociation heats in the ITC cell over an equilibrium concentration range of ~0.2
– 2.4 µM for the sEGFRWT:EGF complex. Because the heat of injection does not change
32
is so low (and can not be distinguished from instrumental noise), we conclude that the ∆Hdimerization
for the sEGFRWT:EGF complex is <<|2| kcal/mol.
Two independent lines of evidence argue that ligated, monomeric receptor is qualitatively distinct
from one half of a 2:2 EGF:EGFR dimer. If these two species were the same, dimerization of the
two identical halves of the 2:2 EGF:EGFR dimer would lead to their mutual stabilization. In other
words, dimerization would stabilize the ligand-bound conformation, which would in turn be
manifest as an enhancement of ligand binding upon dimerization. As shown above, however, this
does not happen. It seems reasonable to argue instead, from the data outlined above, that
dimerization of a 1:1 EGF:sEGFR complex must compromise ligand binding to each protomer to
some degree. Whereas monomeric, ligated receptor presumably adopts a conformation that
maximizes ligand-receptor interactions, dimerization presumably compromises some of these
interactions, so that ligand-binding and dimerization energies are not additive. In other words, in
order for dimerization to occur, some ligand-receptor contacts must be compromised. We would
therefore expect that the domain II conformation would be different (possibly very subtly) in the
context of a 1:1 EGF:EGFR complex and a 2:2 EGF:EGFR dimer. Second, the enthalpic
signatures for EGF and TGFα binding differ significantly for sEGFRWT and sEGFRY251A/R285S (Fig.
6 and Table 1). Because dimerization does not contribute significantly to the measured
enthalpies (see Fig. 7B), the difference in ∆H of ~2kcal/mol between sEGFR and the
Y251A/R285S variant can be directly attributed to differences in ligand-binding. The best
explanation for this difference in ∆Hligand-binding is that the ligand:receptor interaction is qualitatively
(and thermodynamically) distinct for monomeric and dimeric receptor. KD values are very similar
(64nM and 41nM respectively), so G differs by less than 0.23 kcal/mol. Yet, H for EGFR
binding differs by ~2 kcal/mol, being enthalpically less unfavorable in the case where dimerization
is possible. These observations argue that G for EGF binding to sEGFR dimers remains