Atomic and Electronic Structures of Hydrated Polymolybdates
by First Principles Calculations
A. Togo
1;*, I. Tanaka
1, K. Murase
1, T. Yamamoto
2, T. Suga
2and E. Matsubara
31
Department of Materials Science and Engineering, Kyoto University, Kyoto 606-8501, Japan 2Fukui Institute for Fundamental Chemistry, Kyoto University, Kyoto 606-8103, Japan 3Institute for Materials Research, Tohoku University, Sendai 980-8577, Japan
First principles calculations of hydrated polymolybdates complexes have been made with an atomic orbital basis molecular orbital method. Hydration effects are taken into account by the conductor-like screening model (COSMO) using dielectric constant of water. Hydrated heptapolymolybdate, Mo7O246, shows a low symmetry structure, which agrees well to experimental results,i.e., X-ray diffraction of crystalline
salts and X-ray absorption fine structure of the hydrated complex. Contrary to that, the hydrated heteropolymolybdate, NiMo6O2410prefer to
exhibit the high symmetry structure. Inspection of the electronic states found that the Ni ion exhibits trivalent state ord7configuration in a
formal sense. Jahn-Teller distortion around Ni is therefore evident. Such distortion cannot be found in CrMo6O249or CoMo6O249.
(Received February 27, 2004; Accepted March 30, 2004)
Keywords: first priciples calculation, conductor-like screening model, polymolybdates
1. Introduction
Hexavalent molybdenum species, which are dissolving as orthomolybdate ions MoO42 in basic aqueous media, are known to be condensed into a series of cluster ions called iso-polymolybdates, e.g. heptamolybdate Mo7O246 and octa-molybdate Mo8O264, in concert with their protonation. The structure of the polymolybdate ions have been deter-mined traditionally with X-ray analyses of their crystalline salts, such as M6(Mo7O24)xH2O (Mþ = Naþ, Kþ, NH4þ,
etc.).
Recently, atomic structure of Mo7O246 ion in aqueous solutions were experimentally studiedin-situby combing X-ray absorption spectroscopy (XAS), anomalous X-X-ray scat-tering (AXS), and small-angle X-ray scatscat-tering (SAXS) by the present authors.1,2) The study showed that the local
structure of Mo7O246(aq) is almost the same as that of its salt crystals such as (NH4)6(Mo7O24)4H2O.3) They are composed of distorted MoO6octahedrons with three different Mo-O bond lengths ranging from 0.17 to 0.23 nm. The structure of the distorted octahedron is similar to that can be found in MoO3 crystal.4)
In the presence of second metal ions, molybdate ions form another series of complex polymolybdates,e.g.AlMo6O249 and CoMo6O249, which are called heteropolymolybdates. Recently, Muraseet al.have reported the formation of heter-opolymolybdate involving Ni2þions at around pH 5 using the factor analysis, a multivariate analysis, of visible absorption spectra for the Ni2þ-MoO
42 system.5) Assessed from the spectral change, the heteropolymolybdate had the composi-tion NiMo6O2410. Since the salts of NiMo6O2410 have not been prepared successfully, the structure of NiMo6O2410 ion has not been clarified by means of X-ray diffraction. Ac-cording to the analysis of XAFS (X-ray absorption fine struc-ture) for the Ni2þ-MoO
42solution, however, the present au-thors have found the configuration of NiMo6O2410ion to be one Ni2þion surrounded by six sets of distorted MoO
6 octa-hedron with three different Mo-O bond lengths.
Since aqueous Ni2þ-MoO
42 solutions are promising electrolytic baths for Ni-Mo alloy electroplating, which is an alternative to hard chromium coatings, the molybdate species therein bare not only scientific interest but also application potential. In the present study, we made first principles calculations to elucidate the atomic structure of these polymolybdates in aqueous solutions. Hydration effects are taken into account by the conductor-like screening model (COSMO)6,7) using dielectric constant of water. Firstly we examine the structure of the heptapolymolybdate, Mo7O246. Then we examine the structure of the hydrated heteropoly-molybdates, MMo6O249(M = Cr, Co and Ni) with special interests on the role of the alloying element on the structure.
2. Computational Procedure
Calculations were made using a first principles molecular orbital method. A program code DMOL38,9) has been
adopted. It uses numerical basis functions as values on an atomic-centered spherical-polar mesh. The radial part of the basis functions were obtained by solving the atomic Kohn-Sham equations numerically. Greater variational freedom was obtained by doubling the basis-set size, namely by generation of an entire second set of functions. A 3d orbital was also included for oxygen. The basis sets are called DND (double numerical plus d-functions) that is equivalent to 6-31G* basis sets used in Gaussian code.10) Generalized
gradient corrected functional (GGA)11) was used to treat
exchange and correlation potential. Spin polarization was taken into account only for molecules. Geometry optimiza-tion was truncated when the maximum displacement was smaller than 0.5 pm. Hydration effects were included by COSMO, which will be explained in detail in the next chapter.
3. Effects of Hydration on the Atomic Arrangements
When a charged molecule or solute is present in water, it is surrounded by a shell of highly ordered water molecules, which is known as hydration. The phenomenon can be *Graduated Student, Kyoto University
Special Issue on Grain Boundaries, Interfaces, Defects and Localized Quantum Structures in Ceramics
described by a charged species embedded in a dielectric medium from the macroscopic viewpoint. The medium or solvent polarized at the surface of the cavity that accom-modates the charged solute. COSMO is a popular approach to include the solvation effects in quantum mechanical calcu-lations. Its usefulness to accurately reproduce geometry and energetics of solvated molecules has been well confirmed.
The effect of hydration on the optimized geometry can be clearly demonstrated in a complicated molecule. In a simple system, such as in MoO42, neither optimized geometry nor electronic structure was significantly modified by applying the hydration effect. It simply translated energy eigenvalues because of the compensation of the extra charge in a molecule.
As an example of a complicated molecule, we have examined structures of a Mo(VI)-citrate complex MoO3 -(cit)4, (cit4= C
6H4O74) with and without the hydration. The dielectric constant of water,"¼78:4was used. In order to examine the hydration effects, other computational conditions such as basis functions and initial structure for geometry optimization were kept to be identical. Figure 1 shows structures with and without the hydration after geometrical optimization. Two structures look quite different especially around the Mo ion. When the hydration effect is taken into account, Mo is six fold coordinated. The Mo-O bond lengths are 180 pm3, 205 pm, 233 pm and 250 pm. The atomic arrangement is analogous to that of the anion structure of MoO3(cit)4 in a crystal K4[MoO3(cit)] 2H2O reported in Ref. 12. The Mo-O bond lengths in the crystal are 173, 174, 176, 205, 224, and 241 pm. On the other hand, one of the Mo-O bonds is broken when the hydration effects are not taken into account. The structure without the hydration is far from that of the crystal. The need of the hydration effects on the structural optimization of such a complicated molecule can be clearly demonstrated. COSMO is confirmed to be a quite successful approach to include the effect.
4. Geometry and Charge Distribution of Hydrated Heptapolymolybdate, Mo7O246
It has been widely accepted that atomic structure of the Mo7O246(aq) is basically the same as that in its salt crystal. It shows approximatelyC2vpoint symmetry, which is rather
low. As will be mentioned in the next chapter, some heteropolymolybdates MMo6O24n with transition metal
ions such as Cr3þ and Co3þ are known to exhibit higher point symmetryD3dwith the alloying element at the center of
the cluster. Figure 2 shows two kinds of geometry with the same composition of Mo7O246. Calculations of these two clusters have been conducted in order to examine their difference in energy and charge distribution. The low symmetry structure shows lower total energy than that of the high symmetry structure by 0.84 eV. The result clearly implies energetical preference of the lower symmetry structure, which well agrees to the experimental result. Bond distances in these two clusters are summarized in Table 1. The Mo-O bond lengths of both of the structures can be classified into three groups with 177–180 pm, 192–201 pm and 219–240 pm. This is analogous to the presence of three kinds of the same number of Mo-O bonds in the crystal of MoO3that are 170, 194 and 229 pm. The number of the three kinds of bonds is the same for the two structures. The major difference between the two structures is the spatial distribu-tion of the bonds. All of seven MoO66 octahedrons in the low symmetry structure have three pairs of different Mo-O bond lengths. On the other hand, the central MoO66 octahedron is almost regular in the high symmetry structure. All of Mo-O bonds in the octahedron have the same length of 196 pm. Only the O-Mo-O angle is about 4 deviated from that in the regular octahedron,i.e., 90. The higher energy of the high symmetry structure should be ascribed to the presence of the nearly regular octahedron which is forced to be formed due to the high symmetry.
with hydration without hydration O
H C Mo
Fig. 1 Theoretically optimized structures of MoO3(cit)4with and without
the hydration.
M
O2 O3 M
O1
O1' O2''
O2 O1'
high symmetry low symmetry
O2' O2'
O1 Mo1
Mo2
Mo1 Mo2
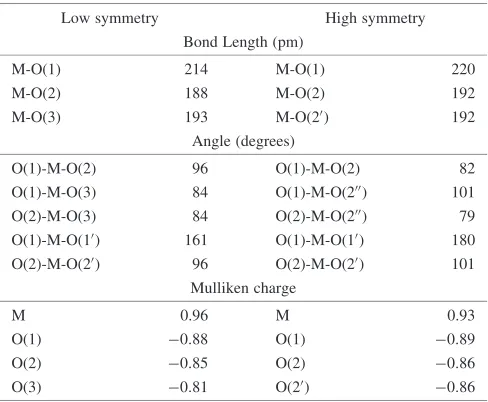
[image:2.595.307.548.76.201.2]Fig. 2 High and low symmetry structures of MMo6O24n.
Table 1 Bond lengths of Mo-O and O-M-O angle by the present calculations in high and low symmetry Mo7O246 in comparison to
experimental data of (NH4)6(Mo7O24)4H2O.
Low symmetry High symmetry Calculation Experiment Calculation
Bond length (pm)
M-O(1) 192 M-O(1) 189 M-O(1) 196 M-O(2) 180 M-O(2) 174 M-O(2) 196 M-O(3) 222 M-O(3) 225 M-O(200) 196
Angle (degree)
O(1)-M-O(2) 100 O(1)-M-O(2) 101 O(1)-M-O(2) 86 O(1)-M-O(3) 78 O(1)-M-O(3) 77 O(1)-M-O(200) 94
O(2)-M-O(3) 84 O(2)-M-O(3) 83 O(2)-M-O(200) 86
O(1)-M-O(10) 147 O(1)-M-O(10) 143 O(1)-M-O(10) 180
[image:2.595.305.549.280.430.2] [image:2.595.56.282.641.757.2]The structure of the hydrated Mo7O246 complex opti-mized in the present study agrees well with that in the salt crystal such as (NH4)6(Mo7O24)4H2O as can be found in Table 1. Difference in Mo-O distances in the two complexes is smaller than 6%.In-situXAFS study by Shinodaet al.1,2)
found that the Mo-O distances of the hydrated Mo7O246 complex can be classified into three groups,i.e., 173, 193 and 224 nm. The number of bonds in each group is the same. The present theoretical results are consistent to the XAFS results, too.
Mulliken charge of Mo and O atoms are summarized in Table 2. The cationic charge is larger in the central Mo than that of the outer Mo in both structures. However, the difference is much larger in the high symmetry structure. The Mulliken charge of Mo in the hydrated MoO42molecule as discussed in the previous chapter is +0.82, which is significantly lower than that in the present hydrated hepta-polymolybdate, Mo7O246, although the formal charges of Mo in two molecules are the same as +6.
5. Structure of Heteropolymolybdate, NiMo6O2410 (aq)
Structure of the heteropolymolybdate, NiMo6O2410(aq) is also examined with two different point symmetries fixing the position of the Ni ion at the center of the complex. In contrast to the hydrated heptapolymolybdate, Mo7O246, we found that the high symmetry structure shows 1.24 eV lower energy than the low symmetry structure. The Mo-O distances and Mullilen charges are summarized in Table 3. It is interesting that the NiO610 octahedron located at the center of the complex is elongated from the regular one. Such distortion cannot be observed in the MoO66octahedron located at the center of the hydrated high symmetry Mo7O246as described in the previous chapter.
Major results by the Mulliken’s population analysis of the spin-polarized calculation are two fold: 1) The Ni ion shows magnetic moment ofþ0:75. Since the Mulliken charge of the Ni isþ0:93, 81% of Ni charges are those with majority spin. This means that Ni ion is spin-polarized in formal sense. 2) Small magnetic moments are induced on Mo ions, although formal charge of Mo(VI) isd0. They are ranging from 0.14 to 0.16. The moments on Ni and Mo are antiferromagnetically coupled. Spin structures are qualitatively the same in the low symmetry structure.
Since Ni(II) hasd8electrons and a fully occupiedegband
with the majority spin, there is no energy benefit associated with the elongation of an octahedron which is often observed for those octahedral complexes with partially occupied eg
band such as Cu(II). In other words, there is no Jahn-Teller type energy benefit in Ni(II) complexes with octahedral symmetry when it is spin-polarized. Some Ni(II) complexes such as [Ni(CN)4]2 are known to adopt a square planar geometry. But they do not have magnetic moment. Some other factors should be taken into account to explain the distortion of the NiMo6O2410(aq). Its electronic structure is therefore analyzed.
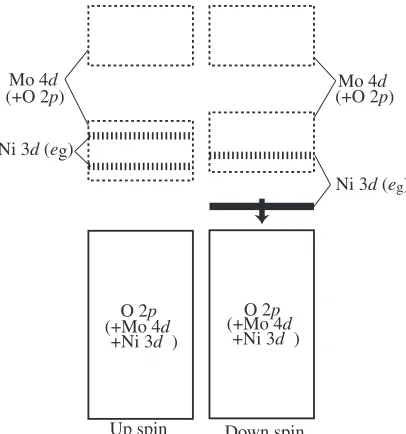
The density of states for NiMo6O2410(aq) near HOMO (highest occupied molecular orbital) and LUMO (lowest unoccupied molecular orbital) is shown in Fig. 3. It looks complicated, which arises not merely from the size of the system but from the mixture of Mo-4dand O-2porbitals with Ni-3d orbitals. Both of HOMO and LUMO are mainly composed of Mo-4d mixed with O-2p orbitals. Molecular orbital energies of some of these Mo-4d + O-2p type molecular orbitals are located close to the Ni-3d bands with majority and minority spins. It is very interesting to note that the Ni-eg orbital splits into two with only one occupied
orbital. This means that the Ni ion is ind7configuration as in Ni(III), not in d8 configuration as in Ni(II). The electronic structure is schematically drawn in Fig. 3. The distortion can therefore be clearly explained by the presence of the Ni(III) with partially occupiedegð#Þorbital which can exhibit Jahn-Teller distortion.
In the present calculation, the number of electrons have been chosen assuming Ni(II), Mo(VI) and O(II). Never-theless, the computational result show Ni(III), not Ni(II). This means that Mo6O2412 has more electron affinity than Ni(II) in the given environment. In the following chapter, we show results of NiMo6O249(aq) on the basis of assumption of Ni(III) in order to examine the difference between NiMo6O2410(aq) and NiMo6O249(aq).
6. Structures of Heteropolymolybdate, MMo6O249 (aq),
(M = Cr, Co, Ni)
[image:3.595.46.291.86.184.2]Crystal structures of salts with CrMo6O249(aq) and CoMo6O249(aq) ions have been reported in literature. But
Table 2 Mulliken charges of atoms in high and low symmetry Mo7O246.
Mulliken charge
low symmetry high symmetry
M 2.23 M 2.42
O(1) 0:89 O(1) 0:90
O(2) 0:82 O(2) 0:90
O(3) 0:93 O(200) 0:90
[image:3.595.305.549.93.294.2]Mo(1) 1.94 Mo(1) 1.91 Mo(2) 1.89 Mo(2) 1.91
Table 3 Bond lengths of Mo-O and O-M-O angle in high and low symmetry NiMo6O2410complex together with Mulliken charges.
Low symmetry High symmetry Bond Length (pm)
M-O(1) 214 M-O(1) 220
M-O(2) 188 M-O(2) 192
M-O(3) 193 M-O(20) 192
Angle (degrees)
O(1)-M-O(2) 96 O(1)-M-O(2) 82 O(1)-M-O(3) 84 O(1)-M-O(200) 101
O(2)-M-O(3) 84 O(2)-M-O(200) 79
O(1)-M-O(10) 161 O(1)-M-O(10) 180
O(2)-M-O(20) 96 O(2)-M-O(20) 101
Mulliken charge
M 0.96 M 0.93
O(1) 0:88 O(1) 0:89
O(2) 0:85 O(2) 0:86
that for M = Ni has not been reported in the authors’ best knowledge. Cr(III) and Co(III) ared3 andd6 configurations in a formal sense. They are thus expected not to show the Jahn-Teller distortion assuming that Co(III) is in the low spin configuration. As a mater of fact, their crystals do not show such distortion. The computed results are compared to the experimental results on salt crystals in Table 4. The agree-ments are satisfactory. Co ion does not show magnetic moments and therefore it is in the low spin state. It is worthwhile to note that the O-M-O angle is slightly deviated from that of the regular octahedron similar to the case of Mo7O246(aq) as described previously. Certainly the
[image:4.595.65.271.76.327.2]dis-tortion cannot be ascribed to the Jahn-Teller disdis-tortion. Computed results of NiMo6O249(aq) as shown together in Table 4 are qualitatively the same as that of
NiMo-6O2410(aq). Ni ion shows electronic configuration of Ni(III) ord7displaying Jahn-Teller distortion. A small distortion of the O-Ni-O angle is also evident in this case.
7. Summary
First principles calculations of hydrated polymolybdates complexes have been made with an atomic orbital basis molecular orbital method with GGA. Major results can be summarized as follows:
(1) Hydration effects on the structure of a MoO3(cit)4, (cit4 = C
6H4O74) complex can be well taken into account by COSMO using dielectric constant of water. (2) Energies of hydrated heptapolymolybdate, Mo7O246, with low and high symmetry structures were compared. The low symmetry structure is found to be preferable by 0.84 eV. The presence of a nearly regular octahedron that is forced to be present in the high symmetry structure is concluded as the origin of the hindrance of the high symmetry structure of Mo7O246.
(3) Contrary to that, the hydrated heteropolymomolybdate, NiMo6O2410, is found to prefer the high symmetry structure. It is more stable than the low symmetry structure by 1.24 eV. The Ni atom in the complex is spin-polarized with antiferromagnetic coupling to the surrounding Mo atoms. It exhibits Ni(III) configuration in a formal sense and displays Jahn-Teller distortion. (4) Other heteropolymomolybdate with Cr(III) and Co(III)
are present instead of Ni(III) do not show such a distortion, because they are in d3 and d6 (low spin) configurations, respectively.
4 3 2 1 0 1 2 3 4 -8 -6 -4 -2 0 2 4
HOMO
Up spin Down spin
Ni 3d
Mo 4d O 2p
Ni 3d (eg) Ni 3d (eg) Ni 3d (eg)
Ni 3d (eg)
Ener
gy / eV
Partial density of states / states.atom-1.eV-1
Ni 3d (eg)
Up spin Down spin
O 2p O 2p
(+Mo 4d +Ni 3d )
(+O 2Mo 4p) (+O 2p)
d Mo 4d
Ni 3d (eg)
(+Mo 4d +Ni 3d )
[image:4.595.302.550.95.343.2] [image:4.595.68.271.366.583.2]Fig. 3 Partial density of states of selected orbitals for the optimized high symmetry Ni-Mo complex. Its schematic view is shown in a bottom panel.
Table 4 Theoretical bond lengths and O-M-O angle in MMo6O249
(M=Cr, Co, Ni) in comparison to experimental data of salt crystals. Bond length (pm) Angle (degrees) Calculation Experiment Calculation Experiment
CrO6O249
M-O(1) 201 M-O(1) 197 O(1)-M-O(2) 81 O(1)-M-O(2) 84 M-O(2) 201 M-O(2) 197 O(1)-M-O(200) 99 O(1)-M-O(200) 95
M-O(200) 201 M-O(200) 197 O(2)-M-O(200) 81 O(2)-M-O(200) 85
O(1)-M-O(10) 180 O(1)-M-O(10) 180
O(2)-M-O(20) 99 O(2)-M-O(20) 95
CoO6O249
M-O(1) 196 M-O(1) 190 O(1)-M-O(2) 81 O(1)-M-O(2) 85 M-O(2) 196 M-O(2) 192 O(1)-M-O(200) 99 O(1)-M-O(200) 95
M-O(200) 201 M-O(200) 197 O(2)-M-O(200) 81 O(2)-M-O(200) 84
O(1)-M-O(10) 180 O(1)-M-O(10) 180
O(2)-M-O(20) 99 O(2)-M-O(20) 96
NiMo6O249
M-O(1) 221 O(1)-M-O(2) 80 M-O(2) 191 O(1)-M-O(200) 100
M-O(200) 191 O(2)-M-O(200) 82
O(1)-M-O(10) 180
Acknowledgements
This work was supported by the Grant-in-Aid for Scientific Research on Priority Areas (No. 751) from the Ministry of Education, Culture, Sports, Science and Technology of Japan, and the computational materials science unit in Kyoto University.
REFERENCES
1) K. Shinoda and E. Matsubara: Hyomen Gijutsu49(1998) 87–93 (in Japanese).
2) K. Shinoda, E. Matsubara, M. Saito, Y. Waseda, T. Hirato, Y. Awakura: Z. Naturforsch A52(1997) 855–862.
3) P. Leveret, B. M. Gatehouseand and H. T. Evans jr: J. Chem. Soc.,
Dalton Trans. (1972–1975) 505–514. 4) L. Kihlborg: Ark. Kemi21(1963) 357–364.
5) K. Murase, H. Ando, E. Matsubara, T. Hirato and Y. Awakura: J. Electrochemi. Soc.147(6) (2000) 2210–2217.
6) A. Klamt and G. Schuurmann: J. Chem. Soc. Perkin Trans.2(1993) 799–805.
7) J. Andzelm and Ch. Kolmel and A. Klamt: J. Chem. Phys.103(1995) 9312–9320.
8) B. Delly: J. Chem. Phys.92(1990) 508–517. 9) B. Delly: J. Chem. Phys.94(1991) 7245–7250.
10) Gaussian is a code name of popular first principles program. See the web site at http://www.gaussian.com.
11) J. P. Perdew, J. A. Chevary, S. H. Vosko, K. A. Jackson, M. R. Pederson, D. J. Singh and C. Fiolhais: Phys. Rev. B46(1992) 6671– 6687.