Ethyl 6-methyl-2-oxo-4-[4-(1
H
-tetrazol-
5-yl)phenyl]-1,2,3,4-tetrahydro-
pyrimidine-5-carboxylate–dimethyl-formamide–water (2/1/1)
Hua-Yong Ouyang,a* Yi-Qi Changband Lu Zhaob
aDepartment of Chemical Engineering, Nanjing College of Chemical Technology, Nanjing 210048, People’s Republic of China, andbDepartment of Applied Chemistry, Nanjing College of Chemical Technology, Nanjing 210048, People’s Republic of China
Correspondence e-mail: ouyanghuay@126.com
Received 19 November 2013; accepted 26 November 2013
Key indicators: single-crystal X-ray study;T= 293 K; mean(C–C) = 0.003 A˚; Rfactor = 0.059;wRfactor = 0.157; data-to-parameter ratio = 16.4.
The asymmetric unit of the title compound, 2C15H16N6O3 -C3H7NOH2O, contains two independent ethyl 6-methyl-2-oxo-4-[4-(1H -tetrazol-5-yl)phenyl]-1,2,3,4-tetrahydropyrimid-ine-5-carboxylate molecules, in which the dihedral angles between the tetrazole and benzene rings are 20.54 (12) and 12.13 (12). An intramolecular C—H O hydrogen bond occurs in each molecule. In the crystal, N—H O, N—H N, O—H O and O—H N hydrogen bonds, as well as weak C—H O and C—H N hydrogen bonds, link the molecules into a three-dimensional supramolecular architecture. – stacking is also observed between parallel tetrazole rings of adjacent molecules, the centroid–centroid distance being 3.482 (6) A˚ .
Related literature
For applications of hydropyrimidine derivatives and related compounds, see: Atwalet al.(1990); Kappe & Stadler (2004).
Experimental
Crystal data
2C15H16N6O3C3H7NOH2O Mr= 747.79
Triclinic,P1 a= 10.198 (2) A˚ b= 13.262 (3) A˚ c= 13.771 (3) A˚
= 81.14 (3)
= 73.32 (3)
= 81.14 (3)
V= 1750.9 (6) A˚3 Z= 2
MoKradiation
= 0.11 mm1 T= 293 K
0.400.300.20 mm
Data collection
Rigaku Mercury2 diffractometer Absorption correction: multi-scan
(CrystalClear; Rigaku, 2005) Tmin= 0.832,Tmax= 1.000
18496 measured reflections 7997 independent reflections 5573 reflections withI> 2(I) Rint= 0.037
Refinement
R[F2> 2(F2)] = 0.059 wR(F2) = 0.157 S= 1.12 7997 reflections
487 parameters
H-atom parameters constrained max= 0.38 e A˚
3 min=0.29 e A˚
[image:1.610.313.566.546.675.2]3
Table 1
Hydrogen-bond geometry (A˚ ,).
D—H A D—H H A D A D—H A
N5—H5A O1W 0.86 1.80 2.646 (3) 170 N9—H9A O7 0.86 1.82 2.683 (3) 176 N10—H10B O2 0.86 1.98 2.801 (2) 160 N11—H11B N2i
0.86 2.26 3.014 (3) 147 N12—H12A O1 0.86 2.04 2.884 (2) 165 N13—H13B O7ii
0.86 2.44 3.176 (3) 144 O1W—H1WA N8ii
0.85 2.17 2.985 (3) 160 O1W—H1WB O1iii 0.85 2.01 2.677 (2) 135 C5—H5D O3 0.96 2.07 2.817 (3) 133 C16—H16A O5 0.96 2.03 2.781 (3) 133 C26—H26A O7 0.93 2.59 3.431 (3) 151 C32—H32A N3iv
0.96 2.53 3.463 (4) 164 C33—H33A N3iv
1.00 2.53 3.506 (3) 164
Symmetry codes: (i)x;yþ1;zþ1; (ii)xþ1;y;zþ1; (iii)x;y;z1; (iv)
xþ1;y1;zþ1.
Data collection: CrystalClear (Rigaku, 2005); cell refinement:
CrystalClear; data reduction:CrystalClear; program(s) used to solve structure:SHELXS97(Sheldrick, 2008); program(s) used to refine
Acta Crystallographica Section E Structure Reports Online
structure: SHELXL97 (Sheldrick, 2008); molecular graphics:
SHELXTL(Sheldrick, 2008); software used to prepare material for publication:SHELXTL.
Supplementary data and figures for this paper are available from the IUCr electronic archives (Reference: XU5754).
References
Atwal, K. S., Rovnyak, G. C., Schwartz, J., Moreland, S., Hedberg, A., Gougoutas, J. Z., Malley, M. F. & Floyd, D. M. (1990).J. Med. Chem.A33, 1510–1515.
supporting information
Acta Cryst. (2014). E70, o1–o2 [https://doi.org/10.1107/S1600536813032224]
Ethyl 6-methyl-2-oxo-4-[4-(1
H
-tetrazol-5-yl)phenyl]-1,2,3,4-tetrahydro-pyrimidine-5-carboxylate
–
dimethylformamide
–
water (2/1/1)
Hua-Yong Ouyang, Yi-Qi Chang and Lu Zhao
S1. Comment
3,4-Dihydropyimidin-2(1H)-ones have shown good drug activity (Atwal et al., 1990; Kappe & Stadler, 2004). The tetrazoles have been showed that analogs of biologically active carboxylic acids in which the carboxyl group is replaced
by a 5-tetrazolyl group might interfere with the normal utilization of the respective carboxylic acids. The Biginelli
derivative was obtained from p-cyanobenzaldehyde, that was used to yield tetrazole derivative. Here we report the
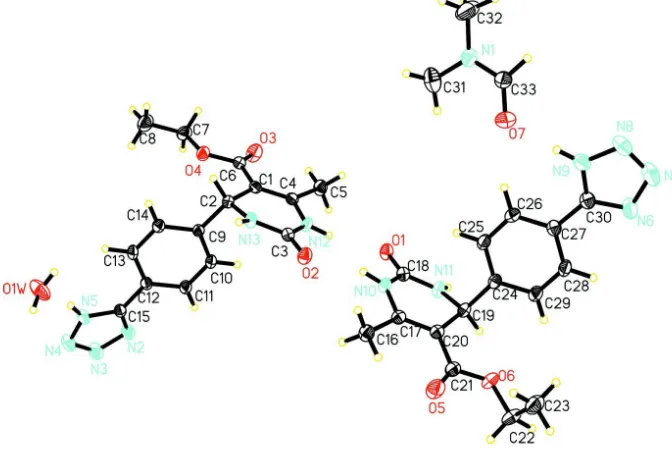
synthesis and crystal structure of the title compound (Fig. 1).
The bond distances and bond angles in the title compound agree very well with the corresponding distances and angles
reported for a closely related compound. There are two biginelli derivatives and two solvate molecules in an asymmetric
unit. The inter-molecular N–H···O and C–H···O hydrogen bonds link the compound to two-dimensional structure (Table
1), in which they may be effective in the stabilization of the structure. π-π contact between the tetrazole rings, Cg1-Cg1i
[symmetry code: (i) -x, 1-y, -z, where Cg1 and Cg1i are centroids of the rings (N2-C15)] may further stabilize the
structure, centroid-centroid distance of 3.482 (6) Å.
S2. Experimental
Cyanobenzaldehyd and ethyl acetoacetate and urea (1:1:1) was added to round-bottom flask without solvent under
nitro-gen. The temperature was raised to 80°C in one hour gradually and the mixture was stirred at this temperature for 12 h.
The system was treated with 30 ml of ethanol 95% and cooled. The precipitate was filtered and washed with a small
amount of ethanol 95%. The above-mentioned compound (10 mmol) was added to sodium azide (15 mmol) and ammonia
chloride (12 mmol) with DMF solvent. The temperature was raised to 115°C in one hour gradually and the mixture was
stirred at this temperature for 36 h. The system was treated with 30 ml of water and cooled. The precipitate was filtered at
pH 3. Single crystals suitable for X-ray diffraction analysis were obtained from slow evaporation of a solution of the title
compound in DMF/water at room temperature
S3. Refinement
H-atoms bonded to the C-atoms were positioned geometrically and refined using a riding model with C—H = 0.93–1.00
Å and Uiso(H) = 1.5Ueq(C) for methyl H atoms and 1.2Ueq(C) for the others. H-atoms bonded to the N-atoms and O-atom
were located from a difference Fourier map and refined in riding mode with O—H = 0.85 and N—H 0.86 Å, Uiso(H) =
Figure 1
Perspective structure of the title compound, showing the atom-numbering scheme.
Figure 2
[image:4.610.139.474.343.624.2]Ethyl 6-methyl-2-oxo-4-[4-(1H-tetrazol-5-yl)phenyl]-1,2,3,4-tetrahydropyrimidine-5-carboxylate– dimethylformamide–water (2/1/1)
Crystal data
2C15H16N6O3·C3H7NO·H2O
Mr = 747.79
Triclinic, P1 Hall symbol: -P 1
a = 10.198 (2) Å
b = 13.262 (3) Å
c = 13.771 (3) Å
α = 81.14 (3)°
β = 73.32 (3)°
γ = 81.14 (3)°
V = 1750.9 (6) Å3
Z = 2
F(000) = 788
Dx = 1.418 Mg m−3
Mo Kα radiation, λ = 0.71073 Å Cell parameters from 7997 reflections
θ = 2.6–27.5°
µ = 0.11 mm−1
T = 293 K Prism, colorless 0.40 × 0.30 × 0.20 mm
Data collection
Rigaku Mercury2 diffractometer
Radiation source: fine-focus sealed tube Graphite monochromator
Detector resolution: 13.6612 pixels mm-1
CCD_Profile_fitting scans Absorption correction: multi-scan
(CrystalClear; Rigaku, 2005)
Tmin = 0.832, Tmax = 1.000
18496 measured reflections 7997 independent reflections 5573 reflections with I > 2σ(I)
Rint = 0.037
θmax = 27.5°, θmin = 3.1°
h = −13→13
k = −17→17
l = −17→17
Refinement
Refinement on F2
Least-squares matrix: full
R[F2 > 2σ(F2)] = 0.059
wR(F2) = 0.157
S = 1.12 7997 reflections 487 parameters 0 restraints
Primary atom site location: structure-invariant direct methods
Secondary atom site location: difference Fourier map
Hydrogen site location: inferred from neighbouring sites
H-atom parameters constrained
w = 1/[σ2(F
o2) + (0.0633P)2 + 0.4646P]
where P = (Fo2 + 2Fc2)/3
(Δ/σ)max = 0.011
Δρmax = 0.38 e Å−3
Δρmin = −0.29 e Å−3
Special details
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes.
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2,
conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used
only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2
are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger.
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
x y z Uiso*/Ueq
Atomic displacement parameters (Å2)
U11 U22 U33 U12 U13 U23
O1 0.0282 (8) 0.0599 (10) 0.0366 (8) −0.0082 (7) −0.0067 (6) −0.0106 (7) O2 0.0329 (8) 0.0580 (10) 0.0408 (8) −0.0118 (7) −0.0081 (7) −0.0110 (7) O3 0.0405 (10) 0.0812 (13) 0.0524 (10) −0.0202 (9) 0.0033 (8) −0.0228 (9) O4 0.0410 (9) 0.0513 (9) 0.0401 (8) −0.0168 (7) −0.0134 (7) −0.0043 (7) O5 0.0564 (12) 0.0963 (15) 0.0508 (11) −0.0395 (11) 0.0006 (9) −0.0144 (10) O6 0.0430 (9) 0.0453 (9) 0.0578 (10) −0.0151 (7) −0.0123 (8) −0.0157 (8) O7 0.0874 (14) 0.0418 (10) 0.0728 (13) −0.0136 (9) −0.0276 (11) −0.0024 (9) O1W 0.0929 (15) 0.0643 (12) 0.0405 (10) 0.0199 (11) 0.0065 (10) 0.0026 (9) C5 0.0467 (13) 0.0597 (15) 0.0323 (11) −0.0137 (11) −0.0028 (10) −0.0115 (10)
Geometric parameters (Å, º)
C1—C4 1.331 (3) C22—O6 1.438 (3)
C1—C6 1.449 (3) C22—C23 1.479 (4)
C1—C2 1.494 (3) C22—H22A 0.9700
C2—N13 1.448 (2) C22—H22B 0.9700
C2—C9 1.503 (3) C23—H23A 0.9600
C2—H2A 0.9800 C23—H23B 0.9600
C3—O2 1.219 (2) C23—H23C 0.9600
C3—N13 1.323 (3) C24—C25 1.370 (3)
C3—N12 1.346 (3) C24—C29 1.371 (3)
C4—N12 1.367 (3) C25—C26 1.363 (3)
C4—C5 1.477 (3) C25—H25A 0.9300
C6—O3 1.187 (3) C26—C27 1.368 (3)
C6—O4 1.331 (2) C26—H26A 0.9300
C7—O4 1.428 (3) C27—C28 1.367 (3)
C7—C8 1.480 (4) C27—C30 1.448 (3)
C7—H7A 0.9700 C28—C29 1.352 (3)
C7—H7B 0.9700 C28—H28A 0.9300
C8—H8A 0.9600 C29—H29A 0.9300
C8—H8B 0.9600 C30—N6 1.306 (3)
C8—H8C 0.9600 C30—N9 1.317 (3)
C9—C14 1.367 (3) C31—N1 1.433 (3)
C9—C10 1.370 (3) C31—H31A 0.9600
C10—C11 1.355 (3) C31—H31B 0.9600
C10—H10A 0.9300 C31—H31C 0.9600
C11—C12 1.371 (3) C32—N1 1.433 (4)
C11—H11A 0.9300 C32—H32A 0.9600
C12—C13 1.367 (3) C32—H32B 0.9600
C12—C15 1.447 (3) C32—H32C 0.9600
C13—C14 1.361 (3) C33—O7 1.212 (3)
C13—H13A 0.9300 C33—N1 1.293 (3)
C14—H14A 0.9300 C33—H33A 1.0042
C15—N2 1.302 (3) N2—N3 1.344 (3)
C15—N5 1.315 (3) N3—N4 1.279 (3)
C16—C17 1.476 (3) N4—N5 1.323 (3)
C16—H16A 0.9600 N5—H5A 0.8600
C16—H16C 0.9600 N7—N8 1.274 (3)
C17—C20 1.331 (3) N8—N9 1.333 (3)
C17—N10 1.366 (3) N9—H9A 0.8600
C18—O1 1.225 (2) N10—H10B 0.8600
C18—N11 1.317 (3) N11—H11B 0.8600
C18—N10 1.346 (3) N12—H12A 0.8600
C19—N11 1.441 (3) N13—H13B 0.8600
C19—C20 1.504 (3) O1W—H1WA 0.8499
C19—C24 1.509 (3) O1W—H1WB 0.8500
C19—H19A 0.9800 C5—H5D 0.9601
C20—C21 1.448 (3) C5—H5E 0.9601
C21—O5 1.185 (3) C5—H5B 0.9599
C21—O6 1.330 (3)
C11—C10—H10A 119.5 N1—C32—H32A 109.5 C9—C10—H10A 119.5 N1—C32—H32B 109.5 C10—C11—C12 120.31 (19) H32A—C32—H32B 109.5 C10—C11—H11A 119.8 N1—C32—H32C 109.5 C12—C11—H11A 119.8 H32A—C32—H32C 109.5 C13—C12—C11 119.11 (19) H32B—C32—H32C 109.5 C13—C12—C15 121.13 (18) O7—C33—N1 124.6 (3) C11—C12—C15 119.75 (18) O7—C33—H33A 126.2 C14—C13—C12 120.26 (19) N1—C33—H33A 109.1 C14—C13—H13A 119.9 C33—N1—C31 120.3 (2) C12—C13—H13A 119.9 C33—N1—C32 122.1 (2) C13—C14—C9 120.81 (19) C31—N1—C32 117.6 (2) C13—C14—H14A 119.6 C15—N2—N3 105.71 (18) C9—C14—H14A 119.6 N4—N3—N2 110.67 (18) N2—C15—N5 108.38 (18) N3—N4—N5 106.21 (18) N2—C15—C12 126.34 (19) C15—N5—N4 109.04 (19) N5—C15—C12 125.27 (19) C15—N5—H5A 125.5 C17—C16—H16A 109.5 N4—N5—H5A 125.5 C17—C16—H16B 109.5 C30—N6—N7 106.4 (2) H16A—C16—H16B 109.5 N8—N7—N6 110.6 (2) C17—C16—H16C 109.5 N7—N8—N9 106.5 (2) H16A—C16—H16C 109.5 C30—N9—N8 108.6 (2) H16B—C16—H16C 109.5 C30—N9—H9A 125.7 C20—C17—N10 119.30 (19) N8—N9—H9A 125.7 C20—C17—C16 127.56 (19) C18—N10—C17 123.34 (17) N10—C17—C16 113.14 (18) C18—N10—H10B 118.3 O1—C18—N11 123.67 (19) C17—N10—H10B 118.3 O1—C18—N10 120.24 (18) C18—N11—C19 124.76 (17) N11—C18—N10 116.09 (18) C18—N11—H11B 117.6 N11—C19—C20 108.71 (16) C19—N11—H11B 117.6 N11—C19—C24 111.60 (17) C3—N12—C4 123.98 (17) C20—C19—C24 112.44 (17) C3—N12—H12A 118.0 N11—C19—H19A 108.0 C4—N12—H12A 118.0 C20—C19—H19A 108.0 C3—N13—C2 125.87 (17) C24—C19—H19A 108.0 C3—N13—H13B 117.1 C17—C20—C21 120.85 (19) C2—N13—H13B 117.1 C17—C20—C19 119.56 (18) C6—O4—C7 115.84 (17) C21—C20—C19 119.60 (18) C21—O6—C22 115.97 (19) O5—C21—O6 122.2 (2) H1WA—O1W—H1WB 109.5 O5—C21—C20 126.7 (2) C4—C5—H5D 109.5 O6—C21—C20 111.16 (19) C4—C5—H5E 109.4 O6—C22—C23 111.0 (2) H5D—C5—H5E 109.5
O6—C22—H22A 109.4 C4—C5—H5B 109.5
Hydrogen-bond geometry (Å, º)
D—H···A D—H H···A D···A D—H···A
N5—H5A···O1W 0.86 1.80 2.646 (3) 170 N9—H9A···O7 0.86 1.82 2.683 (3) 176 N10—H10B···O2 0.86 1.98 2.801 (2) 160 N11—H11B···N2i 0.86 2.26 3.014 (3) 147
N12—H12A···O1 0.86 2.04 2.884 (2) 165 N13—H13B···O7ii 0.86 2.44 3.176 (3) 144
O1W—H1WA···N8ii 0.85 2.17 2.985 (3) 160
O1W—H1WB···O1iii 0.85 2.01 2.677 (2) 135
C5—H5D···O3 0.96 2.07 2.817 (3) 133 C16—H16A···O5 0.96 2.03 2.781 (3) 133 C26—H26A···O7 0.93 2.59 3.431 (3) 151 C32—H32A···N3iv 0.96 2.53 3.463 (4) 164
C33—H33A···N3iv 1.00 2.53 3.506 (3) 164