organic papers
o218
Srdjan Milovacet al. C14H16Cl2N2O2 DOI: 101107/S160053680100232X Acta Cryst.(2001). E57, o218±o219 Acta Crystallographica Section EStructure Reports
Online ISSN 1600-5368
5-Chloro-6-nitroso-2-norbornene dimer as a motif for
supramolecular assembly
Srdjan Milovac,a* Vesna SÆimunicÂ-MezÏnaricÂ,aHrvoj VancÏik,aAleksandar VisÏnjevacb and Biserka KojicÂ-ProdicÂb
aDepartment of Chemistry, Faculty of Science,
University of Zagreb, Strossmayerov trg 14, HR-10000 Zagreb, Croatia, andbRudjer BosÏkovicÂ
Institute, PO Box 180, HR-10002 Zagreb, Croatia
Correspondence e-mail: aleksandar.visnjevac@irb.hr
Key indicators
Single-crystal X-ray study
T= 100 K
Mean(C±C) = 0.003 AÊ
Rfactor = 0.030
wRfactor = 0.083
Data-to-parameter ratio = 10.3
For details of how these key indicators were automatically derived from the article, see http://journals.iucr.org/e.
#2001 International Union of Crystallography Printed in Great Britain ± all rights reserved
The title compound, 1,2-bis(5-chloronorbornen-6-yl)diazene 1,2-dioxide, C14H16Cl2N2O2, has a crystallographic centre of symmetry. The norbornane cages aretrans-oriented and the nitroso groups are in positions 3 and 6. This compound might be used as a starting point for the construction of two different polymeric structures due to the photochromism observed in the solid state.
Comment
Recently, we found that the monomer±dimer equilibrium of C-nitroso compounds could be used as a system for supra-molecular self-assembly (see below, monomer±dimer equili-brium of C-nitroso compounds), because the azodioxide dimer affords photochromism in the solid state (VancÏiket al., 2001). Basic nitroso monomeric units convenient for the construction of supramolecular structures should have a rigid carbon skeleton with two azo groups which serve as chemical recep-tors oriented in proper spatial positions.
Here we propose the polymeric structures that may have nitroso groups on 2- and (MOTIF 1) or on 3- and 6-(MOTIF 2) positions of the norbornane skeleton.
Possibilities of such structures depend on the stereo-chemistry of the basic motif, such as the title structure, (I) (see below), which has only one azodioxide group. The C C double bond on each norbornene unit is a functionality on which additional nitroso groups, necessary for the polymer formation, could be added.
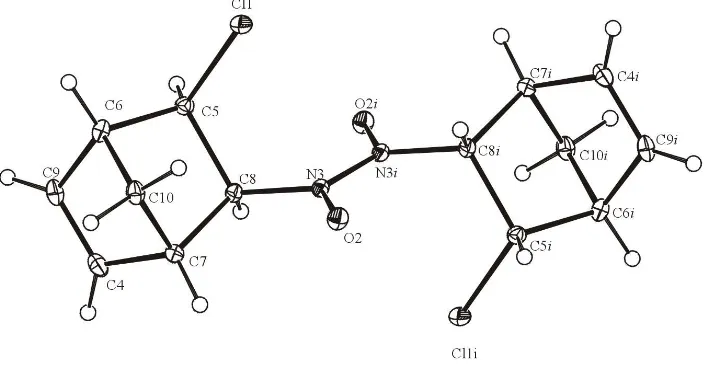
The inversion centre is located at the midpoint of the N N bond which connects two monomeric units into the dimer structure (Fig. 1). The norbornene cages aretrans-oriented.
Experimental
The title compound was prepared by direct addition of NOCl to norbornadiene (Ciattoniet al., 1964; Metzger & Meier, 1971, 1990; Miller, 1961). Crystals of (I) were obtained by evaporation from a dichloromethane solution containing a few drops of tetrahydrofuran at 277 K.
Crystal data
C14H16Cl2N2O2
Mr= 315.19
Monoclinic,P21/n
a= 5.9247 (7) AÊ
b= 13.5925 (11) AÊ
c= 8.3407 (7) AÊ = 95.452 (8)
V= 668.7 (7) AÊ3
Z= 2
Dx= 1.565 Mg mÿ3
MoKradiation Cell parameters from 25
re¯ections = 18.5±25.3 = 0.49 mmÿ1
T= 100 (5) K Prism, colourless 0.180.160.12 mm
Data collection
Enraf±Nonius CAD-4 diffract-ometer
/2scans
Absorption correction: scan (Northet al., 1968)
Tmin= 0.920,Tmax= 0.968
1527 measured re¯ections 1364 independent re¯ections 1076 re¯ections withI> 2(I)
Rint= 0.025
max= 26.3
h=ÿ7!7
k=ÿ16!0
l= 0!10
3 standard re¯ections frequency: 120 min intensity decay: none
Re®nement
Re®nement onF2
R[F2> (F2)] = 0.030
wR(F2) = 0.083
S= 1.02 1364 re¯ections 132 parameters
All H-atom parameters re®ned
w= 1/[2(F
o2) + (0.0407P)2
+ 0.40P]
whereP= (Fo2+ 2Fc2)/3
(/)max= 0.011
max= 0.32 e AÊÿ3
min=ÿ0.24 e AÊÿ3
Extinction correction:SHELXL97 Extinction coef®cient: 0.0055 (12)
Table 1
Selected geometric parameters (AÊ,).
Cl1ÐC5 1.796 (2)
O2ÐN3 1.268 (2) N3ÐC8N3ÐN3i 1.481 (2)1.315 (2)
O2ÐN3ÐC8 123.01 (14) O2ÐN3ÐN3i 120.92 (10)
N3iÐN3ÐC8 116.02 (12)
Cl1ÐC5ÐC6 110.55 (12)
Cl1ÐC5ÐC8 112.90 (11) N3ÐC8ÐC5 113.24 (15) N3ÐC8ÐC7 111.39 (14)
Symmetry code: (i) 1ÿx;1ÿy;1ÿz.
All H-atom positional and Uisoparameters were re®ned. CÐH bond lengths lie between 0.91 (2) and 0.99 (2) AÊ.
Data collection: CAD-4 Software (Enraf±Nonius, 1989); cell re®nement:SET4 andCELDIMinCAD-4Software(Enraf±Nonius, 1989); data reduction:HELENA(Spek, 1997); program(s) used to solve structure: SIR97 (Altomare et al., 1997); program(s) used to re®ne structure:SHELXL97 (Sheldrick, 1997); molecular graphics: ORTEPII (Johnson, 1976); software used to prepare material for publication:PLATON(Spek, 1990).
References
Altomare, A., Cascarano, C., Giacovazzo, C., Guagliardi, A., Moliterni, A. G. G., Burla, M. C., Polidori, G., Camalli, M. & Spagna, R. (1997).SIR97. University of Bari, Italy.
Ciattoni, P., Lorenzini, A. & Gallinella, E. (1964).Chim. Ind.46, 286±291. Enraf±Nonius (1989).CAD-4Software. Version 5.0. Enraf±Nonius, Delft, The
Netherlands.
Johnson, C. K. (1976).ORTEPII. Report ORNL-5138. Oak Ridge National Laboratory, Tennessee, USA.
Metzger, H. & Meier, H. (1971).Methoden der Organischen Chemie, Houben-Weyl, Band 11, pp. 926±938..
Metzger, H. & Meier, H. (1990).Methoden der Organischen Chemie, Houben-Weyl, Band E16a, pp. 958±959.
Miller, J. B. (1961).J. Org. Chem.26, 4905±4907.
North, A. C. T., Phillips, D. C. & Mathews, F. S. (1968).Acta Cryst.A24, 351± 359.
Sheldrick, G. M. (1997).SHELXL97. University of GoÈttingen, Germany. Spek, A. L. (1990).Acta Cryst.A46, C-34.
Spek, A. L. (1997).HELENA.Utrecht University, The Netherlands. VancÏik, H., SÆimunicÂ-MezÏnaricÂ, V., CÏaleta, I., MlinaricÂ-Majerski, K. &
VeljkovicÂ, J. (2001). In preparation.
Figure 1
supporting information
sup-1
Acta Cryst. (2001). E57, o218–o219
supporting information
Acta Cryst. (2001). E57, o218–o219 [doi:10.1107/S160053680100232X]
5-Chloro-6-nitroso-2-norbornene dimer as a motif for supramolecular assembly
Srdjan Milovac, Vesna
Š
imuni
ć
-Me
ž
nari
ć
, Hrvoj Van
č
ik, Aleksandar Vi
š
njevac and Biserka Koji
ć
-Prodi
ć
S1. Comment
Recently, we found that the monomer–dimer equilibrium of C-nitroso compounds could be used as a system for
supramolecular self-assembly (see below, monomer–dimer equilibrium of C-nitroso compounds), because the azodioxyde
dimer affords photochromism in the solid state (Vančik et al., 2001). Basic nitroso monomeric units convenient for the
construction of supramolecular structures should have a rigid carbon skeleton with two azo groups which serve as
chemical receptors oriented in proper spatial positions.
Here we propose the polymeric structures that may have nitroso groups on 2- and 6- (MOTIF 1) or on 3- and 6-
(MOTIF 2) positions of the norbornane skeleton. Possibilities of such structures depend on the stereochemistry of the
basic motif, such as the title structure, (I) (see below), which has only one azodioxide group. The C═C double bond on
each norbornene unit is a functionality on which additional nitroso groups, necessary for the polymer formation, could be
added.
The inversion centre is located in the midpoint of the N═N bond which connects two monomeric units into the dimer
structure (Fig. 1). The norbornene cages are trans-oriented.
S2. Experimental
The title compound was prepared by direct addition of NOCl to norbornadiene (Ciattoni et al., 1964; Metzger et al.,
1971, ibid. 1990; Miller, 1961). Crystals of (I) were obtained by evaporation from a dichloromethane solution containing
a few drops of tetrahydrofuran at 277 K.
S3. Refinement
Figure 1
An ORTEPII (Johnson, 1976) drawing of (I) with the atomic numbering scheme. Displacement ellipsoids are drawn at the
30% probability level.
5-chloro-6-nitroso-2-norbornene dimer or 1,2-bis(5-chloronorbornen-6-yl)diazene 1,2-dioxide
Crystal data C14H16Cl2N2O2 Mr = 315.19 Monoclinic, P21/n Hall symbol: -P 2yn a = 5.9247 (7) Å b = 13.5925 (11) Å c = 8.3407 (7) Å β = 95.452 (8)° V = 668.7 (7) Å3 Z = 2
F(000) = 328 Dx = 1.565 Mg m−3
Mo Kα radiation, λ = 0.71069 Å Cell parameters from 25 reflections θ = 18.5–25.3°
µ = 0.49 mm−1 T = 100 K Prism, colourless 0.18 × 0.16 × 0.12 mm
Data collection Enraf-Nonius CAD-4
diffractometer
Radiation source: X-ray tube Graphite monochromator θ/2θ scans
Absorption correction: ψ scan (North et al., 1968)
Tmin = 0.920, Tmax = 0.968 1527 measured reflections
1364 independent reflections 1076 reflections with I > 2σ(I) Rint = 0.025
θmax = 26.3°, θmin = 2.0° h = −7→7
k = −16→0 l = 0→10
3 standard reflections every 120 min intensity decay: none
Refinement Refinement on F2 Least-squares matrix: full R[F2 > 2σ(F2)] = 0.030 wR(F2) = 0.083 S = 1.02 1364 reflections 132 parameters
0 restraints 0 constraints
All H-atom parameters refined w = 1/[σ2(Fo2) + (0.0407P)2 + 0.4P]
where P = (Fo2 + 2Fc2)/3 (Δ/σ)max = 0.011
supporting information
sup-3
Acta Cryst. (2001). E57, o218–o219
Δρmin = −0.24 e Å−3 Extinction correction: SHELXL97,
Fc*=kFc[1+0.001xFc2λ3/sin(2θ)]-1/4 Extinction coefficient: 0.0055 (12)
Special details
Geometry. Bond distances, angles etc. have been calculated using the rounded fractional coordinates. All e.s.d.'s are estimated from the variances of the (full) variance-covariance matrix. The cell e.s.d.'s are taken into account in the estimation of distances, angles and torsion angles
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger.
Examination of the data with PLATON (Spek, 1990) showed that there was no solvent accesible voids inside the structure.
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
x y z Uiso*/Ueq
Cl1 0.07717 (8) 0.54022 (4) 0.29490 (7) 0.0200 (2) O2 0.3945 (2) 0.39708 (10) 0.58190 (10) 0.0185 (4) N3 0.4637 (3) 0.45571 (11) 0.47899 (10) 0.0142 (4)
C4 0.5147 (3) 0.30110 (15) 0.1130 (2) 0.0206 (6)
C5 0.2828 (3) 0.47482 (14) 0.1910 (2) 0.0144 (5)
C6 0.1749 (3) 0.38287 (15) 0.1062 (2) 0.0176 (5)
C7 0.4425 (3) 0.31681 (14) 0.2817 (2) 0.0176 (5)
C8 0.4774 (3) 0.42849 (14) 0.3083 (2) 0.0144 (5)
C9 0.3574 (3) 0.34093 (15) 0.0093 (2) 0.0195 (6)
C10 0.1834 (3) 0.30986 (15) 0.2465 (2) 0.0181 (5)
H1 0.652 (5) 0.275 (2) 0.090 (3) 0.036 (7)*
H2 0.345 (3) 0.5219 (14) 0.121 (2) 0.004 (4)*
H3 0.030 (4) 0.3962 (16) 0.048 (3) 0.020 (6)*
H4 0.511 (4) 0.2745 (16) 0.364 (2) 0.014 (5)*
H5 0.616 (4) 0.4480 (15) 0.282 (2) 0.013 (5)*
H6 0.358 (4) 0.3484 (17) −0.103 (3) 0.022 (6)*
H7 0.138 (4) 0.2416 (17) 0.216 (3) 0.016 (5)*
H8 0.105 (4) 0.3324 (15) 0.336 (2) 0.012 (5)*
Atomic displacement parameters (Å2)
U11 U22 U33 U12 U13 U23
C10 0.0215 (9) 0.0164 (10) 0.0165 (9) −0.0046 (8) 0.0022 (8) −0.0021 (8)
Geometric parameters (Å, º)
Cl1—C5 1.796 (2) C7—C8 1.545 (3)
O2—N3 1.268 (2) C7—C10 1.538 (3)
N3—C8 1.481 (2) C4—H1 0.92 (3)
N3—N3i 1.315 (2) C5—H2 0.963 (18)
C4—C7 1.524 (3) C6—H3 0.96 (2)
C4—C9 1.325 (3) C7—H4 0.96 (2)
C5—C6 1.544 (3) C8—H5 0.91 (2)
C5—C8 1.571 (3) C9—H6 0.94 (2)
C6—C9 1.522 (3) C10—H7 0.99 (2)
C6—C10 1.531 (3) C10—H8 0.97 (2)
Cl1···N3 2.873 (3) C6···H4ix 3.04 (2)
Cl1···O2i 3.312 (3) C9···H2x 2.851 (18)
Cl1···N3i 3.160 (3) C9···H4ix 2.77 (2)
Cl1···O2ii 3.184 (3) C9···H5 2.996 (19)
Cl1···H5iii 3.00 (2) H1···O2viii 2.75 (3)
Cl1···H8 2.85 (2) H1···H8viii 2.57 (3)
Cl1···H3iv 3.00 (2) H2···C9x 2.851 (18)
O2···C10 3.183 (3) H2···H6x 2.51 (3)
O2···Cl1ii 3.184 (3) H3···Cl1iv 3.00 (2)
O2···Cl1i 3.312 (3) H4···O2 2.61 (2)
O2···C5i 3.096 (3) H4···C6vii 3.04 (2)
O2···H4 2.61 (2) H4···C9vii 2.77 (2)
O2···H6v 2.74 (2) H5···Cl1xi 3.00 (2)
O2···H8 2.69 (2) H5···C9 2.996 (19)
O2···H5i 2.40 (2) H5···O2i 2.40 (2)
O2···H1vi 2.75 (3) H6···O2xii 2.74 (2)
O2···H7vii 2.56 (2) H6···H2x 2.51 (3)
N3···Cl1 2.873 (3) H7···O2ix 2.56 (2)
N3···Cl1i 3.160 (3) H8···Cl1 2.85 (2)
N3···H8 2.88 (2) H8···O2 2.69 (2)
C5···O2i 3.096 (3) H8···N3 2.88 (2)
C10···O2 3.183 (3) H8···C4vi 3.025 (19)
C4···H8viii 3.025 (19) H8···H1vi 2.57 (3)
O2—N3—C8 123.01 (14) Cl1—C5—H2 106.3 (11)
O2—N3—N3i 120.92 (10) C6—C5—H2 115.2 (11)
N3i—N3—C8 116.02 (12) C8—C5—H2 110.1 (11)
C7—C4—C9 107.68 (16) C5—C6—H3 112.9 (13)
Cl1—C5—C6 110.55 (12) C9—C6—H3 116.5 (15)
Cl1—C5—C8 112.90 (11) C10—C6—H3 118.1 (14)
C6—C5—C8 102.00 (15) C4—C7—H4 116.2 (12)
C5—C6—C9 105.21 (15) C8—C7—H4 116.5 (12)
supporting information
sup-5
Acta Cryst. (2001). E57, o218–o219
C9—C6—C10 100.95 (15) N3—C8—H5 107.0 (11)
C4—C7—C8 102.99 (14) C5—C8—H5 111.0 (12)
C4—C7—C10 100.24 (13) C7—C8—H5 111.3 (13)
C8—C7—C10 101.84 (15) C4—C9—H6 128.6 (15)
N3—C8—C5 113.24 (15) C6—C9—H6 123.9 (14)
N3—C8—C7 111.39 (14) C6—C10—H7 114.9 (14)
C5—C8—C7 102.95 (14) C6—C10—H8 113.7 (12)
C4—C9—C6 107.29 (15) C7—C10—H7 110.3 (14)
C6—C10—C7 93.77 (14) C7—C10—H8 112.2 (13)
C7—C4—H1 125.1 (16) H7—C10—H8 110.9 (19)
C9—C4—H1 126.8 (16)
O2—N3—C8—C5 104.52 (19) C6—C5—C8—N3 −123.96 (15)
O2—N3—C8—C7 −10.9 (2) C6—C5—C8—C7 −3.56 (16)
N3i—N3—C8—C5 −78.0 (2) C8—C5—C6—C10 39.13 (16)
N3i—N3—C8—C7 166.55 (15) Cl1—C5—C8—N3 −5.3 (2)
O2—N3—N3i—O2i 180.0 (4) C5—C6—C10—C7 −58.46 (15)
O2—N3—N3i—C8i −2.4 (3) C5—C6—C9—C4 72.25 (19)
C8—N3—N3i—O2i 2.4 (3) C10—C6—C9—C4 −32.6 (2)
C8—N3—N3i—C8i −180.00 (15) C9—C6—C10—C7 49.61 (16)
C9—C4—C7—C10 33.6 (2) C4—C7—C8—C5 70.61 (16)
C7—C4—C9—C6 −0.8 (2) C10—C7—C8—N3 88.69 (16)
C9—C4—C7—C8 −71.17 (19) C4—C7—C10—C6 −49.76 (16)
C8—C5—C6—C9 −65.59 (16) C8—C7—C10—C6 55.98 (15)
Cl1—C5—C6—C9 174.11 (12) C10—C7—C8—C5 −32.97 (16)
Cl1—C5—C6—C10 −81.18 (14) C4—C7—C8—N3 −167.74 (14)
Cl1—C5—C8—C7 115.09 (13)
Symmetry codes: (i) −x+1, −y+1, −z+1; (ii) −x, −y+1, −z+1; (iii) x−1, y, z; (iv) −x, −y+1, −z; (v) x, y, z+1; (vi) x−1/2, −y+1/2, z+1/2; (vii) x+1/2, −y+1/2,