A Novel Mutation in the Anion Exchanger 1 Gene Is Associated With
Familial Distal Renal Tubular Acidosis and Nephrocalcinosis
Lara Cheidde, MD, MSc*; Teresa Cristina Vieira, MD, PhD‡; Paulo Roberto Moura Lima, PhD§; Sara Teresinha Ollala Saad, MD, PhD§; and Ita Pfeferman Heilberg, MD, PhD*
ABSTRACT. Objective. The anion exchanger gene
(AE1) or band 3 encodes a chloride-bicarbonate (Clⴚ/
HCO3ⴚ) exchanger expressed in the erythrocyte and in the renal␣-intercalated cells involved in urine acidification. The purpose of the present study was to screen for mu-tations in theAE1gene in 2 brothers (10 and 15 years of age) with familial distal renal tubular acidosis (dRTA), nephrocalcinosis, and failure to thrive.
Methods. AE1 mutations were screened by single-strand conformation polymorphism, cloning, and se-quencing.
Results. A complete form of dRTA was confirmed in the 2 affected brothers and an incomplete form in their father. All 3 were heterozygous for a novel 20-bp deletion in exon 20 of theAE1gene. This deletion resulted in 1 mutation in codon 888 (Ala-8883Leu) followed by a premature termination codon at position 889, truncating the protein by 23 amino acids. As band 3 deficiency might lead to spherocytic hemolytic anemia or ovalocy-tosis, erythrocyte abnormalities were also investigated, but no morphologic changes in erythrocyte membrane were found and the osmotic fragility test was normal.
Conclusions. A novel mutation in theAE1gene was identified in association with autosomal dominant dRTA. We suggest that RTA be considered a diagnostic possibility in all children with failure to thrive and nephrocalcinosis.Pediatrics2003;112:1361–1367;distal re-nal tubular acidosis, nephrocalcinosis, anion exchanger 1, band 3.
ABBREVIATIONS. NC, nephrocalcinosis; dRTA, distal renal tu-bular acidosis; CA II, cytoplasmic carbonic anhydrase II; uRBP, urinary retinol binding protein; SSCP, single-strand conformation polymorphism; PCR, polymerase chain reaction.
N
ephrocalcinosis (NC) is defined by calcium deposition within the renal parenchyma. NC can be divided into medullary and cortical forms, the former being the most common one. The main causes of NC in pediatric patients are long-term furosemide treatment in neonates1,2 and renaltubular acidosis during childhood.3Distal renal
tu-bular acidosis (dRTA) is a clinical syndrome identi-fied by hyperchloremic metabolic acidosis secondary to a selective defect in distal renal acidification and characterized by inappropriately high urine pH, hy-pokalemia, and reduced net acid excretion. Rare cases of hereditary dRTA have been described.4 –10
Primary hereditary forms of dRTA are predomi-nantly seen as autosomal dominant traits.4,5,11–13An
autosomal recessive mode of inheritance has also been described in association or not with sensorineu-ral deafness.9,10,13 Some patients with autosomal
dominant dRTA remain asymptomatic until adoles-cence or adulthood, whereas others and those with recessive disease may be severely affected in infancy, with impaired growth and early NC eventually lead-ing to renal insufficiency.4,13
The physiology of distal tubular function is com-plex, and different hypotheses explaining the occur-rence of dRTA have been proposed.5,9,10,13–15
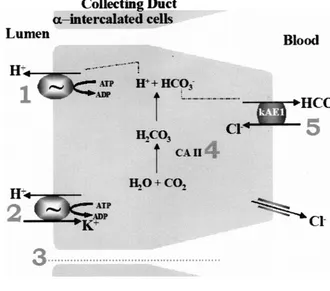
Cyto-plasmic carbonic anhydrase II (CA II) catalyzes the hydration of carbon dioxide to carbonic acid, which dissociates to form bicarbonate (HCO3⫺) and
hydro-gen ions (H⫹), the latter being secreted into the tu-bule through the action of H⫹-ATPase or H⫹/K⫹ -ATPase on the apical membrane of the␣-intercalated cells in the renal collecting duct. The HCO3⫺
gener-ated by this process is transported across the baso-lateral membrane through the anion exchanger 1 (AE1), a HCO3⫺/Cl⫺ anion exchanger. Hypotheses
for the occurrence of dRTA are depicted in Fig 1. One of the possible defects resulting in dRTA may affect the anion exchanger, vital to tubular acid secretion because loss of its function in the presence of con-tinuing H⫹ secretion by the luminal proton pumps would lead to excessive accumulation of HCO3⫺
within the cell, with a consequent reduction in the dissociation of CA II and hence reduced availability of protons for secretion into the tubular lumen.
The members of the AE gene family are widely distributed in tissues, where they are involved in the regulation of intracellular pH and cell volume and in the transcellular transport of acid and base across epithelial cells.16,17 There are at least 4 members of
the AE gene family,18 and the very abundant form
present in red cells (AE1 or band 3) has been stud-ied.11,16,19
Band 3 protein is a 911 amino acid membrane protein encoded by the anion exchanger erythroid isoform (AE1) gene located on chromosome 17q21-qter20 presenting 20 exons21,22 and 3 distinct
func-From the *Nephrology Division, Universidade Federal de Sa˜o Paulo (UNIFESP), Sa˜o Paulo, Brazil; ‡Endocrinology Division, Universidade Fed-eral de Sa˜o Paulo (UNIFESP), Sa˜o Paulo, Brazil; and §Hemocentro, Facul-dade de Cieˆncias Me´dicas de Campinas, UniversiFacul-dade Estadual de Campi-nas (UNICAMP), CampiCampi-nas, Sa˜o Paulo, Brazil.
Received for publication Sep 18, 2002; accepted Mar 16, 2003.
Reprint requests to (I.P.H.) Rua Botucatu, 740 Vila Clementino, Sa˜o Paulo SP, Brazil, CEP 04023–900, Nephrology Division/ Universidade Federal de Sa˜o Paulo. E-mail: ipheilberg@nefro.epm.br
tional domains: 2 cytoplasm tails (N-terminal and C-terminal) and the central integral membrane do-main that has 12 to 14 membrane-spanning regions. The C-terminal tail binds to CA II,23essential for the
anion exchange. The renal isoform of band 3, trun-cated at the N-terminus, described as kidney AE1 (kAE1) lacking erythroid exons 1 to 3, could be pre-dicted to initiate translation at Met 66.19This kAE1 is
localized in the basolateral membrane of the collect-ing duct in␣-intercalated cell.
SeveralAE1mutations have been characterized in association with hereditary spherocytosis14,22,24 –28
and Southeast Asian ovalocytosis.6,8,11 Most of the
studies in the literature have not focused on investi-gating abnormalities in renal acidification because only after 1997 were autosomal dominant and reces-sive forms of dRTA reported to present mutations in theAE1 gene.4,5,10,11,14,15,29 To date, the associations
between dRTA and red blood cell disorders such as spherocytosis or Southeast Asian ovalocytosis have been reported only in the recessive form of dRTA.8,11,14
In the present investigation, we report 2 related patients with hereditary dRTA who were screened for mutations in theAE1 gene. A novel 20-bp dele-tion in exon 20 of theAE1gene was identified.
METHODS Patient Characteristics
Two brothers, 10 and 15 years of age (probands III 2 and III 3 in Fig 2), were referred to the outpatient Renal Stone Clinic of the Nephrology Division, Universidade Federal de Sa˜o Paulo, because of NC and calculus voiding. The mother reported that medical advice was sought for the children at age 3 and 6 years because of failure to thrive. Suspicion of hypophosphatemic rickets was raised, and calcium, phosphorus, and vitamin D supplementation was started at that time for both children. After 4 years of treat-ment, a plain radiography showed severe NC. At the present
admission, in view of the similar signs and symptoms shared by the 2 brothers, a possible hereditary renal disease was considered and a search for a familial disease was started. Written informed consent was obtained from all individuals of the family partici-pating in the study, which was approved by the Ethics Committee of the Universidade Federal de Sa˜o Paulo. Complete metabolic evaluation was performed in the affected brothers and their par-ents only. Complete dRTA was diagnosed in 2 brothers, and incomplete dRTA was diagnosed in their father.
Metabolic Evaluation
The diagnosis of complete dRTA was based on an alkaline pH after an overnight 12-hour fast in the presence of acidosis. Com-plete dRTA is characterized by the inability to lower urinary pH below 5.5 under the stimulus of spontaneous systemic acidemia, whereas in the incomplete form of dRTA despite a high urinary pH, acidemia is lacking as a result of a mechanism of arterial pH compensation. Both forms of dRTA may be clinically indistin-guishable. In individuals with incomplete dRTA, an acute acid
Fig 1. Hypotheses involved in dRTA. Defects in either H⫹/ATPase (1) or
H⫹/K⫹-ATPase (2) on the apical
mem-brane of the H⫹-secreting intercalated
cells of the collecting duct, or an in-creased permeability of this membrane causing excessive back-diffusion of se-creted H⫹ (3), besides defects in
car-bonic anhydrase II (4) orAE1(5) result-ing in failure to establish or maintain a cell-to-lumen hydrogen ion gradient resulting in abnormal renal acidifica-tion.
challenge test is necessary to induce a metabolic acidosis and disclose the failure of lowering urinary pH. The acidification tests, nicely reviewed recently,30can be divided into 3 types: acid
load-ing (ammonium chloride [NH4Cl], calcium chloride, or arginine
hydrochloride), maneuvers that increase the distal delivery of sodium (sodium sulfate infusion and furosemide test), and buffer loading (bicarbonate titration, urine partial pressure of carbon dioxide with urinary alkalinization, and urine partial pressure of carbon dioxide with phosphate infusion). In the present study, the NH4Cl loading (0.1 g or 1.9 mEq/kg body weight), the most
common test, was used. Urine was collected for a period of 3 hours after NH4Cl ingestion, and pH and net acid excretion were
determined.31 A 2-fold increase in ammonium excretion and a
3-fold increase in titratable acidity are considered to be an ade-quate response to the acid load, but the most important and diagnostic parameter was the reduction of urinary pH to⬍5.5 after the load.
The family pedigree is presented in Fig 2. Biochemical analysis was performed only in the affected patients (III 2 and III 3) and their parents (II 2 and II 3) and consisted of the determination of serum calcium, uric acid, creatinine, sodium, potassium, phospho-rus, chloride, bicarbonate, 25(OH)D3, 1,25 (OH)2D3, and blood
smear analysis as well as urinary oxalate, potassium, sodium, calcium, uric acid, citrate, chloride, creatinine, phosphorus, and magnesium determination. Creatinine clearance was also deter-mined and urinary retinol binding protein (uRBP) was measured in spot urine.
Because of the presence of hypercalciuria, NC, and rickets, we suspected a mutation in the chloride channel (CLCN5)32,33 and
thus performed determinations of uRBP as a screening for CLCN5 mutation. Abnormal uRBP excretion was present in both children and their father but not in their mother. Because CLCN5 mutation is a familial tubular syndrome whose inheritance is X-linked re-cessive, with no instances of male-to-male transmission33and the
pattern of inheritance suggested dominant autosomal dRTA trans-mitted by the father, the possibility of a CLCN5 mutation was excluded and theAE1gene was elected as a candidate gene.
DNA Analysis and Polymerase Chain Reaction
Single-strand conformation polymorphism (SSCP) analysis of genomic DNA was performed as previously described.24DNA
was extracted from peripheral blood leukocytes obtained from blood samples using the Puregene DNA Isolation kit (Gentra Systems, Minneapolis, MN). Exons 2 to 20 of theAE1gene were amplified by polymerase chain reaction (PCR) using primers lo-cated on intronic boundaries, as described elsewhere.24The PCR
product of the exon that showed a mobility shift was analyzed by automatic DNA sequencing. The PCR product was subcloned in MAX Efficiency DH5␣Competent Cells (GIBCO-BRL, Rockville, MD) using the Original TA Cloning kit (Invitrogen, Groningen, The Netherlands). The plasmid DNA from positive colonies was isolated using the QIAprep Spin Miniprep kit (QIAGEN, Hilden, Germany). For identifying the mutation in the exons of theAE1
gene observed in the PCR-SSCP analysis, the product was se-quenced by automatic DNA sequencing using the Big Dye reac-tion in the ABI PRISM Model 377 apparatus (Applied Biosystem, Foster City, CA).
RESULTS
On the occasion of the present admission, the di-agnosis of complete dRTA was established for both
children (III 2 and III 3) by the detection of inappro-priately alkaline urinary pH in the presence of spon-taneous metabolic acidosis, as shown in Table 1. The mother (II 2) presented a normal response to the NH4Cl test. Conversely, the father (II 3) maintained an alkaline urinary pH (6.6) and presented a slight but inadequate increase in ammonium excretion and titratable acidity, characterizing an incomplete form of dRTA. Both children had normal renal function, hypokalemia, hypercalciuria, hypocitraturia, hyper-uricosuria, and hyperchloremia, as shown in Table 2. Because serum levels of 25(OH)D3and 1,25 (OH)2D3 were within normal limits, vitamin D supplementa-tion was then withdrawn. The reason for an increase in uRBP in both children and their father remains unclear. Nevertheless, it has already been reported by other investigators in a dRTA setting.34
NC was confirmed by hyperechogenicity of renal pyramids and calcifications in renal ultrasonography as well as parenchymal calcifications by helical com-puted tomography. Pelvicaliceal calcifications were also observed in computed tomography, indicating the association with nephrolithiasis. Potassium citrate sup-plementation was started soon after the diagnosis of dRTA was established. Control of metabolic acidosis was then achieved, and an improvement in growth rate was observed in both children (⬎7 cm/child after 14 months of treatment). Renal calcification has not got worsen; neither did renal function deteriorate during the same period.
The father presented a normal ultrasound but re-ported a history of previous stone voiding and sub-mission to 4 extracorporeal shock wave lithotripsy procedures. The half-sister and the half-brother (pro-bands III 4 and III 1 in Fig 2, respectively) had normal urinary pH (data not shown). The mother had no nephrolithiasis or NC despite the presence of hyper-calciuria (Table 2). All members of the family pre-sented normal renal function, as assessed by serum creatinine levels (data not shown).
Because of hematologic diseases linked to muta-tions in the AE1 gene, a hematologic investigation was initiated. The peripheral blood smears and the osmotic fragility test were normal, indicating no spherocytosis or ovalocytosis. The blood counts in both affected brothers were normal, with the follow-ing values: hematocrit, 45.2% and 42.2%; hemoglo-bin, 15.7 and 14.9 g/dL; mean corpuscular volume, 88 and 90; mean corpuscular hemoglobin
concentra-TABLE 1. Acidification Parameters Under Basal Conditions and After the NH4Cl Load Test
Parents Patients
II 2 (Mother) II 3 (Father) III 2 III 3
Basal Post NH4Cl Basal Post NH4Cl Basal Basal
Blood
pH 7.41 7.27 7.29 7.21 7.26 7.28
HCO3⫺(mmol/L) 25 22 21 24 17 16
Urine
pH 5.9 5.3 6.9 6.6 6.7 6.8
NH4(Eq/min) 28.8 43.1 21.9 35.8 19.7 10.4
HCO3(mmol/L) 10.6 0.4 4.5 2.9 3.3 3.3
tion, 35.0 and 35.3 g/dL; and reticulocyte count, 1.4% and 1.5%, proband III 2 and III 3, respectively.
Genetic Investigation
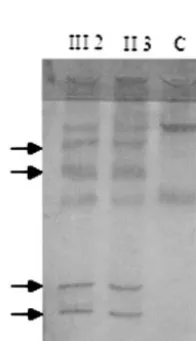
SSCP analysis revealed a mobility shift in exon 20 of theAE1gene in proband III 3 and his father (II 3), as seen in Fig 3. Except for this mobility shift of exon 20, the PCR-SSCP patterns of all other exons, includ-ing intron 3, were normal (data not shown). The 3% agarose gel electrophoresis performed in the entire family (Fig 4) showed a single band of 267 bp in relatives without dRTA and 2 bands (267 bp and 247 bp) in the patients with dRTA (III 2, III 3) and their father (II 3). The smaller band suggested the presence of a deletion in exon 20 of these patients. The PCR product of patient III 3 was subcloned in pCR 2.1, and the product was then sequenced. A 20-bp se-quence starting from the third base in codon 887 was deleted, identifying the AE1 mutation (Fig 5). This
deletion did not alter the protein encoded by codon 887 but resulted in 1 mutation in codon 888 (Ala-8883Leu) followed by a premature termination codon at position 889, truncating the protein by 23 amino acids. Because we evaluated 3 generations and the double band was lacking in the grandpar-ents, we had to confirm parenthood using DNA fin-gerprinting through a technique based on the vari-able number of tandem repeats, VNTR (Profile Kit, Perkin Elmer, Boston, MA). Parenthood was con-firmed, hence suggesting that the deletion resulted from a de novo mutation (Fig 4). We named this mutation Dourados (Band 3 Dourados) in reference to the area of origin of the children.
DISCUSSION
Familial forms of distal RTA have been described since 1968,35 but it was only after 1997 that both
dominant4,5,29 and recessive7,8,11 autosomal forms
were associated withAE1 mutations. In the present investigation, complete dRTA and NC were detected in 2 brothers and incomplete dRTA with nephroli-thiasis in their father. The similar clinical course ob-served in the 2 brothers led us to hypothesize that a genetic cause of the disorder should be sought.
In the present study, the affected brothers and their father shared a 20-bp deletion in exon 20 of the AE1gene or Band 3 (Band 3 Dourados), resulting in a premature termination codon at position 889, trun-cating the protein by 23 amino acids. The deletion at the C-terminal tail was responsible for the double band evidenced by agarose gel electrophoresis of the exon 20 PCR product of the brothers and their father, 1 with 267 and 1 with 247 bp (without 20 bp) instead of the single 267-bp band.
The structure and function of the short C-terminal cytoplasmic tail (33 residues) of band 3 have not been well characterized. Recent studies have proposed that CA II binds toAE1 through the C-terminal tail, catalyzing CO2hydration and favoring HCO3⫺ reab-sorption and H⫹ secretion.23,34,36,37
Fig 3. Screening of mutations in exon 20 of the AE1 gene in patients with dRTA by PCR-SSCP analysis. From left to right, patient III 2 with complete dRTA and NC; his father II 3 with incomplete dRTA and nephrolithiasis; control (C). Arrows indi-cate band shifts in exon 20.
TABLE 2. Serum and Urinary Parameters in the Patients and Their Parents
II 2 II 3 III 2 III 3
Serum
Calcium (mg/dL [mmol/L]) 9.2 (2.30) 9.4 (2.35) 10.1 (2.52) 10.1 (2.52)
Uric acid (mg/dL [mol/L]) 3.2 (190) 6.6 (393) 4.5 (268) 5.0 (297)
Creatinine (mg/dL [mol/L]) 0.8 (71) 0.9 (80) 0.8 (71) 0.7 (62)
Sodium (mEq/L [mmol/L]) ND ND 139 (139) 141 (141)
Potassium (mEq/L [mmol/L]) ND ND 3.1 (3.1) 3.3 (3.3)
Phosphate (mg/dL [mmol/L]) 2.5 (0.81) 2.9 (0.94) 4.9 (1.58) 3.9 (1.26)
Chloride (mEq/L [mmol/L]) 103 (103) 108 (108) 120 (120) 112 (112)
25(OH)D3(ng/mL [ng/mL]) ND ND 16.0 (40) 24.9 (62)
1,25 (OH)2D3(pg/mL [pmol/L]) ND ND 33.3 (87) 29.6 (77)
Urine
Oxalate (mg/d [mol/d]) 34 (378) 36 (400) 29 (322) 18 (200)
Sodium (mEq/d [mmol/d]) 349 (349) 265 (265) 189 (189) 117 (117)
Calcium-mg/kg/d (mmol/kg/d) 4.4 (0.12) 3.0 (0.07) 4.8 (0.12) 10 (0.25)
Uric acid-mg/d (mol/d) 93 (2926) 143 (2159) 285 (2852) 242 (1439)
Citrate-mg/d (mol/d) 474 (2164) 107 (214) 81 (203) 59 (140)
Creatinine-mg/d (mol/d) 1015 (7817) 1430 (4862) 978 (4157) 550 (2210)
RBP mg/L* 0.01 2.47 22.2 20.4
ND indicates not determined.
There are only 3 reported mutations in this C-terminal tail, specifically in exon 20.4,15,38Karet et al4
were the first to identify an intragenic 13-bp dupli-cation in tandem that resulted in a premature termi-nation codon at position 901, truncating the protein in the last 11 amino acids in association with dRTA. Another mutation was Band 3 Vesuvio,18
character-ized by a frameshift deletion in codon 894 resulting in a reading frame for 133 extra codons (instead of 18) before the new stop codon at position 1027,
as-sociated with hereditary spherocytosis but not with dRTA. More recently, Toye et al15reported the
pres-ence of dRTA and NC in 2 brothers who shared the same mutation described by Karet et al4 and an
additional deletion of 9 bp over the sequence that would have coded for amino acids Tyr904-Glu906 of normal band 3, named band 3 Walton.
In the present study, the deletion of 23 amino acids in the mutant AE1 protein probably did not interfere with band 3 insertion into the red cell membrane Fig 4. AE1Mutations in dRTA. Agarose gel
electrophoresis of exon 20 in the entire fam-ily showing a double band (267 and 247 bp) only in patients with dRTA (III 2 and III 3) and their father (patient II 3).
because no hematologic diseases were diagnosed. However, it is possible that the mutant protein may be targeted differently in erythrocytes and kidney cells as already described for other mutations in this region.15 The presence of such mutation in children
who present with dRTA suggests that the extreme C-terminal tail of the band 3 protein must play an important role in anion transport and renal acidifi-cation in the renal tubular cells. Another disturbed mechanism caused by the expression of the mutant band 3 in collecting duct cells may be the lack of proper CA II binding, disturbing the cell capacity for bicarbonate transport, or a decrease in the produc-tion/rapid degradation of mutant mRNA38 coding
forAE1leading to the absence or decrease of net acid movement across renal tubular cells.
Despite that the father and 2 children had the same genetic defect, a minor expressivity resulting in an incomplete form of dRTA was diagnosed in the fa-ther. The incomplete form of the disease probably rendered a milder clinical presentation without hy-percalciuria or bone loss as a result of the lack of persistent systemic acidosis. Besides the phenotypic variation, it is unclear why some patients with dRTA present with stones but no overt NC whereas others display the opposite, as already stated by Karet.13The
possibility that vitamin D treatment, as well as cal-cium and phosphorus supplementation, before the establishment of the dRTA diagnosis might have contributed to aggravate the renal calcifications in the children cannot be excluded.39 – 41 Nevertheless,
as already mentioned earlier, the finding of similar clinical signs and symptoms shared by the 2 brothers suggests that a secondary NC could not represent the single entity presented by these children. The proper control of metabolic acidosis with potassium citrate after the diagnosis of dRTA was established led to an important improvement in growth rate. We suggest that RTA should be considered as a diagnostic pos-sibility in all children with failure to thrive and NC.
REFERENCES
1. Saarela T, Lanning P, Koivisto M, Paavilainen T. Nephrocalcinosis in full-term infants receiving furosemide treatment for congestive heart failure: a study of the incidence and 2-year follow up.Eur J Pediatr. 1999;158:668 – 672
2. Kugelman A, Durand M, Garg M. Pulmonary effect of inhaled furo-semide in ventilated infants with severe bronchopulmonary dysplasia.
Pediatrics. 1997;99:71–75
3. Cremin B, Wiggelinkhuizen J, Bonnici F. Nephrocalcinosis in children.
Br J Radiol. 1982;55:413– 418
4. Karet FE, Gainza FJ, Gyo¨ry AZ, et al. Mutations in the chloride-bicarbonate exchanger geneAE1cause autosomal dominant but not autosomal recessive distal renal tubular acidosis.Proc Natl Acad Sci U S A. 1998;95:6337– 6342
5. Bruce LJ, Cope DL, Jones Gk, et al. Familial distal renal tubular acidosis is associated with mutations in the red cell anion exchanger (band 3,
AE1) gene.J Clin Invest.1997;100:1693–1707
6. Bruce LJ, Wrong O, Toye AM, et al. Band 3 mutations, renal tubular acidosis and South-East Asian ovalocytosis in Malaysia and Papua New Guinea: loss of up to 95% band 3 transport in red cells.Biochem J.
2000;350:41–51
7. Tanphaichitr VS, Sumboonnanonda A, Ideguchi H, et al. NovelAE1
mutations in recessive distal renal tubular acidosis: loss-of-function is rescued by glycophorin A.J Clin Invest1998;102:2173–2179
8. Vasuvattakul S, Yenchitsomanus PT, Vachuanichsanong P, et al. Auto-somal recessive distal renal tubular acidosis associated with Southeast Asian ovalocytosis.Kidney Int.1999; 56:1674 –1682
9. Karet FE, Finberg KE, Nelson RD, et al. Mutations in the gene encoding B1 subunit of H⫹-ATPase cause renal tubular acidosis with
sensorineu-ral deafness.Nat Genet. 1999;21:84 –90
10. Karet FE, Finberg KE, Nayir A, et al. Localization of a gene for autoso-mal recessive distal renal tubular acidosis with norautoso-mal hearing (rdRTA2) to 7q33–34.Am J Hum Genet. 1999;65:1656 –1665
11. Jarolim P, Shayakul C, Prabakaran D, et al. Autosomal dominant distal renal tubular acidosis is associated in three families with heterozygosity for the R589H mutation in theAE1(band 3) Cl2/HCO3exchanger.J Biol Chem. 1998;273:6380 – 6388
12. Rysava´ R, Tesar V, Jirsa M Jr, Brabec V, Jarolim P. Incomplete distal renal tubular acidosis coinherited with a mutation in the band 3 (AE1) gene.Nephrol Dial Transplant. 1997;12:1869 –1873
13. Karet FA. Inherited Distal renal tubular acidosis.J Am Soc Nephrol. 2001;13:2178 –2184
14. Tanner MJA. Band 3 anion exchanger and its involvement in erythro-cyte and kidney disorders.Curr Opin Hematol. 2002;9:133–139 15. Toye AM, Bruce LJ, Unwin RJ, Wrong O, Tanner MJA. Band 3Walton,
a C-terminal deletion associated with distal renal tubular acidosis, is expressed in the red cell membrane but retained internally in kidney cells.Blood. 2001;99:342–347
16. Kopito RR. Molecular biology of the anion exchanger gene family.Int Rev Cytol. 1990;123:177–199
17. Alper SL. The band 3-related anion exchanger (AE) gene family.Annu Rev Physiol. 1991;53:549 –564
18. Vince JW, Carlsson U, Reithmeier RAF. Localization of the Cl-/HCO3
-anion exchanger binding site to the N-terminal region of carbonic anhydrase II.Biochemistry. 2000;39:13344 –13349
19. Tanner MJA. Molecular and cellular biology of the erythrocyte anion exchanger (AE1).Semin Hematol. 1993;30:34 –57
20. Showe LC, Ballantine M, Huebner K. Localization of the gene for the erythroid anion exchange protein, band 3 (EMPB3), to human chromo-some 17.Genomics. 1987;1:71–76
21. Lux SE, John KM, Kopito RR, Lodish HF. Cloning and characterization of band 3, the human erythrocyte anion-exchange protein (AE1).Proc Natl Acad Sci U S A. 1993;86:9089 –9093
22. Tanner MJA. The structure and function of band 3 (AE1). Recent devel-opments.Mol Membr Biol. 1997;14:155–165
23. Vince JW, Reithmeier RAF. Identification of the carbonic anhydrase II binding site in the Cl/HCO3anion exchanger.AE1. Biochemistry. 2000;
39:5527–5533
24. Lima PRM, Gontijo JAR, Faria JBL, Costa FF, Saad STO. Band 3 campinas: a novel splicing mutation in the band 3 gene (AE1) associated with hereditary spherocytosis, hyperactivity of Na⫹/Li⫹
countertrans-port and an abnormal renal bicarbonate handling. Blood. 1997;7: 2810 –2818
25. Jarolim P, Rubin HL, Liu SC, et al. Duplication of 10 nucleotides in the erythroid band 3 (AE1) gene in a kindred with hereditary spherocytosis and band 3 protein deficiency (band 3-Prague).J Clin Invest.1994;93: 121–130
26. Jarolim P, Rubin HL, Brabec V, et al. Mutations of conserved arginines in the membrane domain of erythroid band 3 lead to a decrease in membrane-associated band 3 and to the phenotype of hereditary spherocytosis.Blood. 1995;85:634 – 640
27. Jarolim P, Murray JL, Rubin HL, et al. Characterization of 13 novel band 3 gene defects in hereditary spherocytosis with band 3 deficiency.Blood. 1996;88:4366 – 4374
28. Iolascon A, Miraglia Del Giudice E, Perrota S, Alloisio N, Morle´ L, Delaunay J. Hereditary spherocytosis: from clinical to molecular de-fects.Haematologica.1998;83:240 –257
29. Weber S, Soergel M, Jeck N, Konrad M. Atypical acidosis confirmed by mutation analysis.Pediatr Nephrol. 2000;15:201–204
30. Soriano JR. Renal tubular acidosis: the clinical entity.J Am Soc Nephrol. 2002;13:2160 –2170
31. Lash JP, Arruda JAL. Laboratory evaluation of renal tubular acidosis.
Clin Lab Med. 1993;13:117–129
32. Norden AGW, Scheinman SJ, Dechodt-Lanckman MM, et al. Tubular proteinuria defined by a study of Dent’s (CLCN5 mutation) and other tubular diseases.Kidney Int. 2000;57:240 –249
33. Scheinman SJ. X-linked hypercalciuric nephrolithiasis: clinical syn-dromes and chloride channel mutations.Kidney Int. 1998;53:3–17 34. Reithmeier RAF. A membrane metabolon linking carbonic anhydrase
with chloride/bicarbonate exchangers.Blood Cells Mol Dis. 2001;27: 85– 89
35. Baehner RL, Gilchrist GS, Anderson EJ. Hereditary elliptocytosis and primary renal tubular acidosis in a single family. Am J Dis Child. 1968;115:414 – 419
carboxyl-terminus of human band 3, the erythrocyte Cl-/HCO3- exchanger.J Biol Chem. 1998;273:28430 –28437
37. Perrotta S, Polito F, Cone ML, et al. Hereditary spherocytosis due to a novel frameshift mutation in AE1 cytoplasmic COOH terminal tail: band 3 Vesuvio.Blood. 1999;93:2131–2132
38. Jackson RJ. Cytoplasmatic regulation of mRNA function: the impor-tance of the 3⬘untranslated region.Cell. 1994;74:9 –14
39. Stickler GB, Jowsey J, Bianco Al Jr. Possible detrimental effect of large
doses of vitamin D in familial hypophosphatemic vitamin D resistant rickets.J Pediatr. 1971;79:68 –71
40. Goodyear PR, Kronick JB, Jequier S, Reade TM, Scriver CR. Nephrocal-cinosis and its relationship to treatment of hereditary rickets.J Pediatr. 1987;111:700 –704
41. Reusz GS, Latta K, Hoyer PF, Byrd DJ, Ehrich JHH, Brodehl J. Evidence suggesting hyperoxaluria as a cause of nephrocalcinosis in phosphate treated hypophosphatemic rickets.Lancet. 1990;335:1240 –1243
FETAL PROGRAMMING EXPLAINED?
“Epigenetics may . . . help explain how the seeds of many adult diseases may be planted during fetal life. Studies suggest that the nutrition a fetus receives—as indicated by birth weight—might influence the risk of adult-onset diabetes, heart disease, hypertension and some cancers. The basis for such ‘fetal programming’ has been largely an enigma, but epigenetics may be key. . . . There is no doubt that in the case of the brown or yellow mice, the ‘you are what your mom ate’ phenomenon reflects just such epigenetic influences. . . . Duke scientists fed female mice dietary supplements of vitamin B12, folic acid, betaine and choline just before and throughout their pregnancies. Offspring of mice eating a regular diet had yellowish fur; pups of the supplemented mothers, although genetically identical to the yellow mice, were brown.”
Begley S. Science journal.Wall Street Journal. August 15, 2003
2003;112;1361
Pediatrics
Ollala Saad and Ita Pfeferman Heilberg
Lara Cheidde, Teresa Cristina Vieira, Paulo Roberto Moura Lima, Sara Teresinha
Distal Renal Tubular Acidosis and Nephrocalcinosis
A Novel Mutation in the Anion Exchanger 1 Gene Is Associated With Familial
Services
Updated Information &
http://pediatrics.aappublications.org/content/112/6/1361 including high resolution figures, can be found at:
References
http://pediatrics.aappublications.org/content/112/6/1361#BIBL This article cites 41 articles, 11 of which you can access for free at:
Subspecialty Collections
http://www.aappublications.org/cgi/collection/urology_sub
Urology
following collection(s):
This article, along with others on similar topics, appears in the
Permissions & Licensing
http://www.aappublications.org/site/misc/Permissions.xhtml in its entirety can be found online at:
Information about reproducing this article in parts (figures, tables) or
Reprints
2003;112;1361
Pediatrics
Ollala Saad and Ita Pfeferman Heilberg
Lara Cheidde, Teresa Cristina Vieira, Paulo Roberto Moura Lima, Sara Teresinha
Distal Renal Tubular Acidosis and Nephrocalcinosis
A Novel Mutation in the Anion Exchanger 1 Gene Is Associated With Familial
http://pediatrics.aappublications.org/content/112/6/1361
located on the World Wide Web at:
The online version of this article, along with updated information and services, is
by the American Academy of Pediatrics. All rights reserved. Print ISSN: 1073-0397.