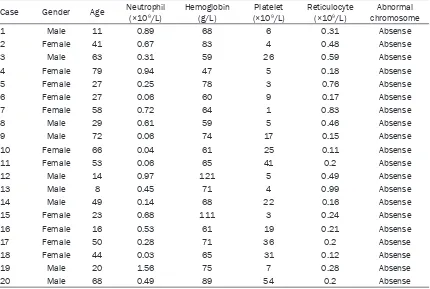
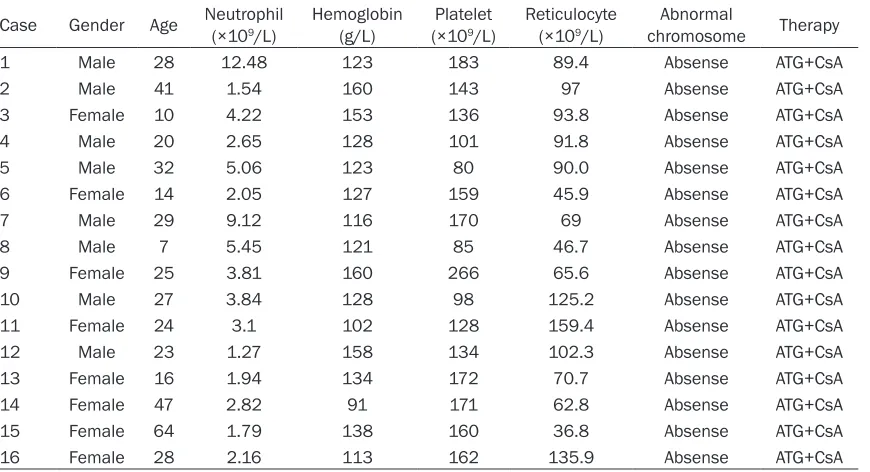
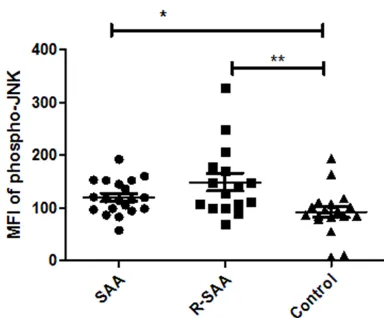
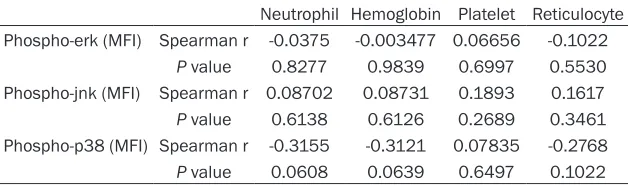
Original Article Activation profile of intracellular mitogen-activated protein kinases in CD8+ T lymphocytes of patients with severe aplastic anemia
8
0
0
Full text
Figure
Related documents