Acta Cryst.(2001). E57, o675±o676 DOI: 10.1107/S1600536801010704 Qi, Zhou, Liu and Chan C8H7FN2O3
o675
organic papers
Acta Crystallographica Section E Structure Reports Online
ISSN 1600-5368
4
000-Fluoro-2
000-nitroacetanilide
Jianying Qi,* Zhongyuan Zhou, Dongsheng Liu and Albert S. C. Chan
Department of Applied Biology and Chemical Technology, The Hong Kong Polytechnic University, Hung Hom, Kowloon, Hong Kong
Correspondence e-mail: [email protected]
Key indicators Single-crystal X-ray study
T= 294 K
Mean(C±C) = 0.002 AÊ
Rfactor = 0.050
wRfactor = 0.168
Data-to-parameter ratio = 15.5
For details of how these key indicators were automatically derived from the article, see http://journals.iucr.org/e.
#2001 International Union of Crystallography Printed in Great Britain ± all rights reserved
The crystal structure of the title compound, C8H7FN2O3,
shows that the amide and nitro groups are rotated slightly out of the aromatic plane, with dihedral angles of 16.30 (6) and 29.60 (10), respectively. The overall molecular organization is
stabilized by well de®ned intermolecular hydrogen bonds that lead to the formation of in®nite chains.
Comment
One of the structural characteristics of 40-¯uoro-20
-nitro-acetanilide, (I), is the presence of a pair of electron-donating (±NH2) and electron-withdrawing (±NO2) groups, a feature
which enhances the dipole moment of this molecule through both inductive and resonance effects (Flettonet al., 1986). In addition, molecule (I) contains a single ¯uoro substituent on its aromatic ring, the signal of which can be easily detectedvia
19F NMR methods. A molecule possessing such structural
characteristics in crystalline form is deemed to be an ideal candidate for examination of the hypothesis of time-reversal symmetry violation, a physics theory postulated in the recent years (Li & Nadin, 1995, 1998). As part of our efforts inves-tigating this theory, we present the crystal structure of (I).
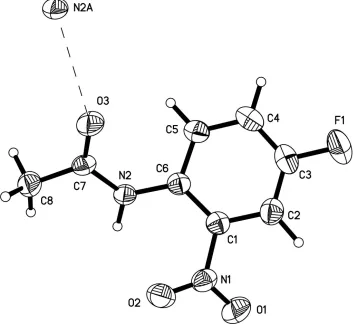
The amide group in (I) (Fig. 1) is rotated out of the ring plane, with a dihedral angle of 16.30 (6). Similarly, the nitro
group is slightly twisted out of the aromatic ring plane by 29.60 (10). The amide N atom approaches the amide O atom
of an adjacent molecule at a distance of 2.9536 (16) AÊ, indi-cating intermolecular hydrogen bonding. It is also noted that (I) crystallizes in a centrosymmetric space group.
According to the theory of time-reversal symmetry viola-tion (Li & Nadin, 1995, 1998), the magnitudes of the two electric currents operating in opposite directions along the same aromatic ring of (I) will be different, thus resulting in two different signals for the chemical shifts of the F atom in (I). Studies into this effect are underway.
Experimental
The title compound was acquired from a commercial source (Aldrich). The crystal used for the data collection was obtained by slow evaporation from acetone±water (2:1) saturated solution at room temperature.
Crystal data C8H7FN2O3
Mr= 198.16
Monoclinic,P21/c
a= 3.9758 (7) AÊ
b= 22.489 (4) AÊ
c= 9.7419 (16) AÊ
= 99.255 (3)
V= 859.7 (3) AÊ3
Z= 4
Dx= 1.531 Mg mÿ3
MoKradiation Cell parameters from 2891
re¯ections
= 1±27.5 = 0.13 mmÿ1
T= 294 (2) K Prism, colorless 0.200.180.16 mm Data collection
CCD area-detector diffractometer
'and!scans
Absorption correction: empirical (SADABS; Sheldrick, 1996)
Tmin= 0.974,Tmax= 0.979
5833 measured re¯ections 1984 independent re¯ections
1284 re¯ections withI> 2(I)
Rint= 0.024 max= 27.6
h=ÿ3!5
k=ÿ28!29
l=ÿ12!12
Re®nement Re®nement onF2
R[F2> 2(F2)] = 0.050
wR(F2) = 0.168
S= 1.06 1978 re¯ections 128 parameters
H-atom parameters constrained
w= 1/[2(F
o2) + (0.1P)2]
whereP= (Fo2+ 2Fc2)/3
(/)max= 0.001 max= 0.36 e AÊÿ3 min=ÿ0.27 e AÊÿ3
Table 1
Hydrogen-bonding geometry (AÊ,).
DÐH A DÐH H A D A DÐH A
N2ÐH2B O3i 0.86 2.10 2.9536 (16) 170
Symmetry code: (i)x;3
2ÿy;12z.
The C-bound H atoms were placed in their geometrically calcu-lated positions and included in the ®nal re®nement in the riding-model approximation.
Data collection: SMART (Siemens, 1995); cell re®nement:
SMART; data reduction: SHELXTL-NT (Siemens, 1995);
program(s) used to solve structure: SHELXS97 (Sheldrick, 1990); program(s) used to re®ne structure:SHELXL97 (Sheldrick, 1997); molecular graphics: SHELXTL-NT; software used to prepare material for publication:SHELXTL-NT.
We thank Dr Tianhu Li for his helpful discussion on the topic of time-reversal symmetry violation.
References
Fletton, R. A., Lancaster, R. W., Harris, R. K., Kenwright, M., Packer, K. J., Waters, D. N. & Yeadon, A. (1986).J. Chem. Soc. Perkin Trans.2, pp. 1075± 1709.
Li, T. & Nadin, A. (1995).Phys.Lett. A,206, 222±224. Li, T. & Nadin, A. (1998).Chirality,10, 289±293. Sheldrick, G. M. (1990).Acta Cryst.A46, 467±473.
Sheldrick, G. M. (1996).SADABS. University of GoÈttingen, Germany. Sheldrick, G. M. (1997).SHELXL97. University of GoÈttingen, Germany. Siemens (1995).SMART(Version 5.0) andSHELXTL-NT(Version 5.10).
Siemens Analytical X-ray Instruments Inc., Madison, Wisconsin, USA.
Figure 1
supporting information
sup-1
Acta Cryst. (2001). E57, o675–o676
supporting information
Acta Cryst. (2001). E57, o675–o676 [doi:10.1107/S1600536801010704]
4
′
-Fluoro-2
′
-nitroacetanilide
Jianying Qi, Zhongyuan Zhou, Dongsheng Liu and Albert S. C. Chan
S1. Comment
One of the structural characteristics of 4′-fluoro-2′-nitroacetanilide, (I), is the presence of a pair of the electron-donating
(–NH2) and electron-withdrawing (–NO2) groups, a feature which enhances the dipole moments of this molecule through
both inductive and resonance effects (Fletton et al., 1986). In addition, molecule (I) contains a single fluoride substituent
on its aromatic ring, the signal of which can be easily detected via 19F NMR methods. A molecule possessing such
structural characteristics in crystalline form is deemed to be an ideal candidate for the examination of the hypothesis of
time-reversal symmetry violation, a physics theory postulated in the recent years (Li & Nadin, 1995, 1998). As part of our
efforts investigating this theory, we present the crystal structure of (I).
The amide group in (I) (Fig. 1) is rotated out of the ring plane, with a dihedral angle of 16.30 (6)°. Similarly, the nitro
group is slightly twisted out of the aromatic ring plane by 29.60 (10)°. The amide N atom approaches the amide O atom
of an adjacent molecule at a distance of 2.9536 (16) Å, indicating intermolecular hydrogen bonding. It is also noted that
(I) crystallizes in a centrosymmetric space group.
According to the theory of time-reversal symmetry violation (Li & Nadin, 1995, 1998), the magnitudes of the two
electric currents operating in opposite directions along the same aromatic ring of (I) will be different, thus resulting in
two different signals for the chemical shifts of F atom in (I). Studies into this effect are underway.
S2. Experimental
The title compound was acquired from a commercial source (Aldrich). The crystal used for the data collection was
obtained by slow evaporation from acetone–water (2:1) saturated solution at room temperature.
S3. Refinement
The C-bound H atoms were placed in their geometrically calculated positions and included in the final refinement in the
Figure 1
The molecular structure of (I) with ellipsoids at the 30% probability level (Bruker, 1995).
4′-fluoro-2′-nitroacetanilide
Crystal data
C8H7FN2O3 Mr = 198.16 Monoclinic, P21/c a = 3.9758 (7) Å b = 22.489 (4) Å c = 9.7419 (16) Å β = 99.255 (3)° V = 859.7 (3) Å3 Z = 4
F(000) = 408
? # Insert any comments here. Dx = 1.531 Mg m−3
Mo Kα radiation, λ = 0.71073 Å Cell parameters from 2891 reflections θ = 1–27.5°
µ = 0.13 mm−1 T = 294 K Prism, colorless 0.20 × 0.18 × 0.16 mm
Data collection
CCD area-detector diffractometer
Radiation source: fine-focus sealed tube Graphite monochromator
φ and ω scans
Absorption correction: empirical (using intensity measurements)
supporting information
sup-3
Acta Cryst. (2001). E57, o675–o676
Rint = 0.024
θmax = 27.6°, θmin = 1.8° h = −3→5
k = −28→29 l = −12→12
Refinement
Refinement on F2 Least-squares matrix: full R[F2 > 2σ(F2)] = 0.050 wR(F2) = 0.168 S = 1.06 1978 reflections 128 parameters 0 restraints
Hydrogen site location: inferred from neighbouring sites
H-atom parameters constrained w = 1/[σ2(Fo2) + (0.1P)2]
where P = (Fo2 + 2Fc2)/3 (Δ/σ)max = 0.001
Δρmax = 0.36 e Å−3 Δρmin = −0.27 e Å−3
Special details
Experimental. ? #Insert any special details here.
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes.
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger.
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
x y z Uiso*/Ueq
F1 −0.1764 (4) 0.48038 (5) 0.13792 (14) 0.0918 (4) O1 0.3856 (5) 0.56838 (6) 0.57282 (13) 0.0834 (5) O2 0.5886 (3) 0.65023 (5) 0.50742 (12) 0.0596 (3) O3 0.1745 (4) 0.74926 (5) 0.10882 (11) 0.0608 (4) N1 0.3966 (4) 0.60772 (6) 0.48671 (13) 0.0494 (3) N2 0.1221 (4) 0.71019 (5) 0.31778 (12) 0.0436 (3)
H2B 0.1282 0.7179 0.4046 0.052*
C1 0.1738 (4) 0.60185 (7) 0.35285 (14) 0.0422 (4) C2 0.0921 (5) 0.54448 (7) 0.30961 (17) 0.0530 (4)
H2A 0.1695 0.5121 0.3653 0.064*
C3 −0.1050 (5) 0.53653 (8) 0.18310 (19) 0.0602 (5) C4 −0.2316 (5) 0.58320 (8) 0.10011 (19) 0.0601 (5)
H4A −0.3654 0.5765 0.0140 0.072*
C5 −0.1565 (5) 0.64031 (7) 0.14688 (17) 0.0497 (4)
H5A −0.2490 0.6722 0.0929 0.060*
C6 0.0549 (4) 0.65141 (7) 0.27332 (14) 0.0404 (4) C7 0.1781 (4) 0.75578 (7) 0.23279 (14) 0.0414 (4) C8 0.2416 (5) 0.81489 (7) 0.30182 (17) 0.0556 (5)
H8D 0.2775 0.8442 0.2339 0.083*
H8A 0.4400 0.8126 0.3722 0.083*
Atomic displacement parameters (Å2)
U11 U22 U33 U12 U13 U23
F1 0.1200 (10) 0.0569 (7) 0.0939 (9) −0.0204 (7) 0.0035 (7) −0.0253 (6) O1 0.1242 (13) 0.0670 (9) 0.0540 (7) 0.0045 (8) −0.0011 (8) 0.0191 (6) O2 0.0580 (8) 0.0673 (7) 0.0502 (7) −0.0008 (6) −0.0010 (6) −0.0042 (6) O3 0.0953 (9) 0.0570 (7) 0.0325 (5) −0.0032 (6) 0.0180 (6) 0.0046 (5) N1 0.0583 (8) 0.0512 (7) 0.0392 (6) 0.0122 (7) 0.0092 (6) 0.0024 (6) N2 0.0619 (8) 0.0423 (7) 0.0268 (5) −0.0009 (6) 0.0073 (5) 0.0000 (5) C1 0.0449 (8) 0.0460 (8) 0.0378 (7) 0.0026 (7) 0.0129 (6) −0.0008 (6) C2 0.0621 (11) 0.0452 (9) 0.0533 (9) 0.0027 (8) 0.0138 (8) 0.0005 (7) C3 0.0670 (12) 0.0508 (10) 0.0650 (10) −0.0113 (9) 0.0173 (9) −0.0154 (8) C4 0.0602 (11) 0.0706 (12) 0.0476 (9) −0.0083 (9) 0.0022 (8) −0.0129 (8) C5 0.0510 (9) 0.0564 (10) 0.0408 (8) −0.0039 (8) 0.0047 (7) −0.0024 (7) C6 0.0442 (8) 0.0455 (8) 0.0336 (7) −0.0004 (6) 0.0122 (6) −0.0013 (6) C7 0.0457 (8) 0.0473 (8) 0.0316 (7) 0.0039 (7) 0.0069 (6) 0.0055 (6) C8 0.0732 (12) 0.0500 (10) 0.0446 (8) −0.0072 (9) 0.0123 (8) 0.0018 (7)
Geometric parameters (Å, º)
F1—C3 1.352 (2) C2—H2A 0.9300
O1—N1 1.2249 (18) C3—C4 1.370 (3)
O2—N1 1.2197 (18) C4—C5 1.379 (2)
O3—C7 1.2144 (17) C4—H4A 0.9300
N1—C1 1.4598 (19) C5—C6 1.398 (2)
N2—C7 1.3585 (18) C5—H5A 0.9300
N2—C6 1.4032 (19) C7—C8 1.493 (2)
N2—H2B 0.8600 C8—H8D 0.9600
C1—C2 1.380 (2) C8—H8A 0.9600
C1—C6 1.396 (2) C8—H8B 0.9600
C2—C3 1.362 (2)
O2—N1—O1 122.90 (14) C5—C4—H4A 120.7
O2—N1—C1 119.49 (13) C4—C5—C6 121.66 (16)
O1—N1—C1 117.59 (14) C4—C5—H5A 119.2
C7—N2—C6 124.37 (12) C6—C5—H5A 119.2
C7—N2—H2B 117.8 C1—C6—C5 116.66 (14)
C6—N2—H2B 117.8 C1—C6—N2 123.38 (13)
C2—C1—C6 122.35 (14) C5—C6—N2 119.87 (13)
C2—C1—N1 115.89 (14) O3—C7—N2 122.56 (14)
C6—C1—N1 121.76 (14) O3—C7—C8 121.97 (14)
C3—C2—C1 118.18 (15) N2—C7—C8 115.46 (12)
C3—C2—H2A 120.9 C7—C8—H8D 109.5
C1—C2—H2A 120.9 C7—C8—H8A 109.5
F1—C3—C4 119.04 (16) H8D—C8—H8A 109.5
F1—C3—C2 118.51 (16) C7—C8—H8B 109.5
C4—C3—C2 122.45 (16) H8D—C8—H8B 109.5
supporting information
sup-5
Acta Cryst. (2001). E57, o675–o676
C3—C4—H4A 120.7
O2—N1—C1—C2 150.10 (15) C2—C1—C6—C5 0.7 (2)
O1—N1—C1—C2 −28.4 (2) N1—C1—C6—C5 179.94 (14)
O2—N1—C1—C6 −29.2 (2) C2—C1—C6—N2 177.08 (15)
O1—N1—C1—C6 152.28 (16) N1—C1—C6—N2 −3.7 (2)
C6—C1—C2—C3 1.6 (3) C4—C5—C6—C1 −2.8 (3)
N1—C1—C2—C3 −177.67 (15) C4—C5—C6—N2 −179.35 (16)
C1—C2—C3—F1 177.73 (16) C7—N2—C6—C1 143.82 (16)
C1—C2—C3—C4 −1.9 (3) C7—N2—C6—C5 −39.9 (2)
F1—C3—C4—C5 −179.78 (18) C6—N2—C7—O3 0.0 (3)
C2—C3—C4—C5 −0.1 (3) C6—N2—C7—C8 179.46 (15)
C3—C4—C5—C6 2.6 (3)
Hydrogen-bond geometry (Å, º)
D—H···A D—H H···A D···A D—H···A
N2—H2B···O3i 0.86 2.10 2.9536 (16) 170