Acta Cryst.(2001). E57, o81±o82 DOI: 101107/S1600536800020730 Philip J. Coxet al. C20H18N2O4
o81
organic papers
Acta Crystallographica Section E
Structure Reports Online
ISSN 1600-5368
Moschamindole
Philip J. Cox,a* Margot
Fergusson,aLutfun Naharband Satyajit D. Sarkera
aSchool of Pharmacy, The Robert Gordon
University, Schoolhill, Aberdeen AB10 1FR, Scotland, andbDepartment of Chemistry,
University of Aberdeen, Meston Walk, Old Aberdeen, Aberdeen AB24 3UE, Scotland
Correspondence e-mail: [email protected]
Key indicators
Single-crystal X-ray study
T= 150 K
Mean(C±C) = 0.005 AÊ
Rfactor = 0.046
wRfactor = 0.097 Data-to-parameter ratio = 7.5
For details of how these key indicators were automatically derived from the article, see http://journals.iucr.org/e.
#2001 International Union of Crystallography Printed in Great Britain ± all rights reserved
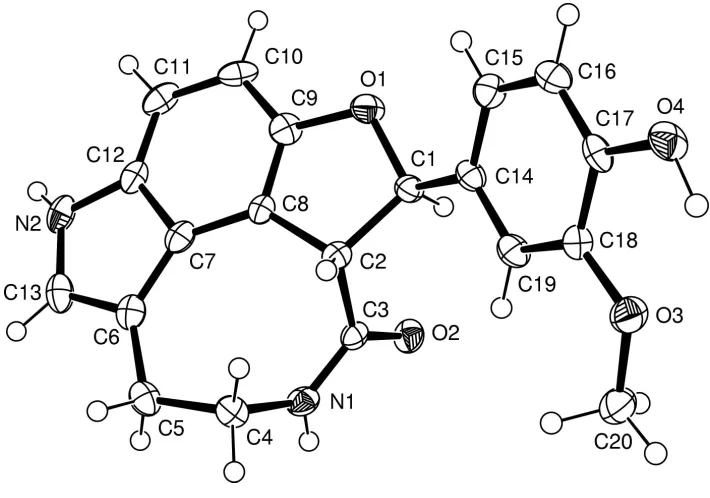
The solid state structure of moschamindole {systematic name: 10-[2-(4-hydroxy-3-methoxyphenyl)]-1,2,3,4,10,10a-hexahy-dro-6H-furo[2,3,4-jk]pyrrolo[4,3,2-ef]-3-benzazocin-1-one}, C20H18N2O4is similar to its solution state structure, previously determined by NMR, and evidence of intermolecular hydrogen bonding is now presented.
Comment
Moschamindole, (I), was obtained as one of four indole alkaloids fromCentaurea moschataand its molecular structure was determined by extensive one-dimensional and two-dimensional NMR spectroscopy (Sarker et al., 1997). We undertook an X-ray study to con®rm the structure determined from NMR data and to establish the presence of hydrogen bonding.
Each H atom (H1 and H2) at the donor N-atom positions (N1 and N2) is involved in an intermolecular hydrogen bond (with O4 and O2, respectively). There is also a weak C11Ð H11 O4 intermolecular hydrogen bond. Three short intra-molecular distances are present where the H A distance `closes' a ®ve-membered ring. Details of these geometries are shown in Table 2. The absolute stereochemistry of the mol-ecule has not been determined but the two chiral centres, C1 and C2, have the same designation (shown as R in Fig. 1). Here, the torsion angle H1ÐC1ÐC2ÐH2 isÿ115 (1). The
eight-membered ring has a conformation where atoms C2, C4, C5, C6, C7 and C8 lie within 0.05 AÊ of a mean plane, with atoms N1 and C3 displaced to the same side of this plane by 1.15 (4) and 1.28 (4) AÊ, respectively. The other four rings adopt planar conformations and the dihedral angle between the aryl ring and the dihydrofuran ring is 43.3 (1). As a result
of ring fusion, valency angle distortions are present around C6, C7 and C8 (Table 1).
Experimental
Moschamindole was obtained from the methanol extract of the seeds ofCentaurea moschata. Crystals for X-ray work were obtained from a methanol solution.
Crystal data
C20H18N2O4
Mr= 350.36 Monoclinic,P21
a= 4.8724 (3) AÊ b= 15.2803 (9) AÊ c= 10.6940 (7) AÊ
= 101.840 (3)
V= 779.25 (8) AÊ3
Z= 2
Dx= 1.493 Mg mÿ3 MoKradiation Cell parameters from 3310
re¯ections
= 2.9±27.5 = 0.11 mmÿ1
T= 150 (2) K Plate, yellow
0.400.200.05 mm
Data collection
Enraf±Nonius KappaCCD area-detector diffractometer
'and!scans to ®ll Ewald sphere 5565 measured re¯ections 1839 independent re¯ections 1272 re¯ections withI> 2(I)
Rint= 0.080
max= 27.5
h=ÿ6!6 k=ÿ19!18 l=ÿ12!13
Re®nement
Re®nement onF2
R[F2> 2(F2)] = 0.046
wR(F2) = 0.097
S= 0.99 1839 re¯ections 245 parameters
H atoms treated by a mixture of independent and constrained re®nement
w= 1/[2(F
o2) + (0.0348P)2] whereP= (Fo2+ 2Fc2)/3 (/)max= 0.007
max= 0.22 e AÊÿ3
min=ÿ0.28 e AÊÿ3
Absolute structure: none
Table 1
Selected geometric parameters (AÊ,). O2ÐC3 1.227 (4) C1ÐC14 1.509 (5) C6ÐC13 1.376 (5)
C7ÐC12 1.422 (5) C8ÐC9 1.367 (4)
C13ÐC6ÐC7 105.5 (3) C13ÐC6ÐC5 122.6 (3) C7ÐC6ÐC5 131.9 (3) C12ÐC7ÐC8 114.9 (3) C12ÐC7ÐC6 107.5 (3)
C8ÐC7ÐC6 137.5 (3) C9ÐC8ÐC7 119.0 (3) C9ÐC8ÐC2 108.7 (3) C7ÐC8ÐC2 132.2 (3)
Table 2
Hydrogen-bonding geometry (AÊ,).
DÐH A DÐH H A D A DÐH A
N1ÐH1 O4i 0.79 (4) 2.29 (4) 3.002 (4) 150 (4)
N2ÐH2 O2ii 0.76 (4) 2.15 (4) 2.909 (4) 171 (3)
O4ÐH4 O3 1.00 (4) 2.17 (3) 2.631 (5) 106 (2) C1ÐH1A O2 1.00 2.27 2.794 (4) 111 C11ÐH11 O4iii 0.95 2.51 3.367 (4) 150
C15ÐH15 O1 0.94 2.37 2.740 (4) 103 Symmetry codes: (i)x;y;zÿ1; (ii) 2ÿx;1
2y;1ÿz; (iii) 2ÿx;12y;2ÿz.
Friedel pairs were merged in the data set, and the absolute con®guration was not determined. H atoms were initially placed in
calculated positions and thereafter allowed to ride on their attached atoms. In the ®nal cycles of least squares, the coordinates of the H atoms attached to N1, N2 and O4 were freely re®ned. Each H atom was given an equivalent Uiso set at 1.2Ueq for its attached atom. Hydrogen-bonding geometries were obtained withPLATON(Spek, 1998).
Data collection: DENZO (Otwinowski & Minor, 1997) and
COLLECT(Hooft, 1998); cell re®nement:DENZOandCOLLECT; data reduction:DENZOandCOLLECT; program(s) used to solve structure:SIR97 (Altomareet al., 1994); program(s) used to re®ne structure: SHELXL97 (Sheldrick, 1997); molecular graphics:
ORTEP-3 (Farrugia, 1997); software used to prepare material for publication:SHELXL97.
We thank the EPSRC for use of the National Crystal-lographic Service, at Southampton University (X-ray data collection) and for the use of the Chemical Database Service at Daresbury (Fletcheret al., 1996).
References
Altomare, A., Cascarano, G., Giacovazzo, C., Guagliardi, A., Burla, M. C., Polidori, G. & Camalli, M. (1994).J. Appl. Cryst.27, 435.
Farrugia, L. J. (1997).J. Appl. Cryst.30, 565.
Fletcher, D. A., McMeeking, R. F. & Parkin, D. (1996).J. Chem. Inf. Comput. Sci.36, 746±749.
Hooft, R. (1998).COLLECT. Nonius BV, Delft, The Netherlands.
Otwinowski, Z. & Minor, W. (1997). Methods Enzymol. 276, 307± 326.
Sarker, S. D., Savchenko, T., Whiting, P., Sik, V. & Dinan, L. N. (1997).Nat. Prod.Lett.9, 189±199.
Sheldrick, G. M. (1997).SHELXL97. University of GoÈttingen, Germany. Spek, A. L. (1998).PLATON.Utrecht University, The Netherlands. Figure 1
supporting information
sup-1
Acta Cryst. (2001). E57, o81–o82
supporting information
Acta Cryst. (2001). E57, o81–o82 [doi:10.1107/S1600536800020730]
Moschamindole
Philip J. Cox, Margot Fergusson, Lutfun Nahar and Satyajit D. Sarker
S1. Comment
Moschamindole, (I), was obtained as one of four indole alkaloids from Centaurea moschata and its molecular structure
was determined by extensive one-dimensional and two-dimensional NMR spectroscopy (Sarker et al., 1997). We
undertook an X-ray study to confirm the structure determined from NMR data and to establish the presence of hydrogen
bonding.
Each H atom (H1 and H2) at the donor N-atom positions (N1 and N2) is involved in an intermolecular hydrogen bond
(with O4 and O2, respectively). There is also a weak C11—H11···O4 intermolecular hydrogen bond. Three short
intramolecular distances are present where the H···A distance `closes′ a five-membered ring. Details of these geometries
are shown in Table 2. The absolute stereochemistry of the molecule has not been determined but the two chiral centres,
C1 and C2, have the same designation (shown as R in Fig. 1). Here, the torsion angle H1—C1—C2—H2 is -115 (1)°. The
eight-membered ring has a conformation where atoms C2, C4, C5, C6, C7 and C8 lie within 0.05 Å of a mean plane, with
atoms N1 and C3 displaced to the same side of this plane by 1.15 (4) and 1.28 (4) Å, respectively. The other four rings
adopt planar conformations and the dihedral angle between the aryl ring and the dihydrofuran ring is 43.3 (1)°. As a result
of ring fusion, valency angle distortions are present around C6, C7 and C8 (Table 1).
S2. Experimental
Moschamindole was obtained from the methanol extract of the seeds of Centaurea moschata. Crystals for X-ray work
were obtained from a methanol solution.
S3. Refinement
Friedel pairs were merged in the data set, and the absolute configuration was not determined. H atoms were initially
placed in calculated positions and thereafter allowed to ride on their attached atoms. In the final cycles of least squares,
the coordinates of the H atoms attached to N1, N2 and O4 were freely refined. Each H atom was given an equivalent Uiso
Figure 1
The atomic arrangement in the title molecule. Displacement ellipsoids are shown at the 50% probability level.
{2-(3-methoxy-4-hydroxyphenyl)}-dihydrofuro[kl]-1H- pyrrolo[fg]-2-oxo-1,2,3,4,5,6-hexahydro-3-benzazocine
Crystal data
C20H18N2O4 Mr = 350.36
Monoclinic, P21 a = 4.8724 (3) Å b = 15.2803 (9) Å c = 10.6940 (7) Å β = 101.840 (3)° V = 779.25 (8) Å3 Z = 2
F(000) = 368 Dx = 1.493 Mg m−3
Mo Kα radiation, λ = 0.71073 Å Cell parameters from 3310 reflections θ = 2.9–27.5°
µ = 0.11 mm−1 T = 150 K Plate, yellow 0.4 × 0.2 × 0.05 mm
Data collection
Enraf Nonius KappaCCD area-detector diffractometer
Radiation source: Enraf Nonius FR591 rotating anode
Graphite monochromator
Detector resolution: 9.091 pixels mm-1 φ and ω scans to fill Ewald sphere 5565 measured reflections
1839 independent reflections 1272 reflections with I > 2σ(I) Rint = 0.080
θmax = 27.5°, θmin = 3.3° h = −6→6
k = −19→18 l = −12→13
Refinement
Refinement on F2 Least-squares matrix: full R[F2 > 2σ(F2)] = 0.046 wR(F2) = 0.097 S = 0.99
245 parameters 1 restraint
Primary atom site location: structure-invariant direct methods
supporting information
sup-3
Acta Cryst. (2001). E57, o81–o82
Hydrogen site location: inferred from neighbouring sites
H atoms treated by a mixture of independent and constrained refinement
w = 1/[σ2(Fo2) + (0.0348P)2] where P = (Fo2 + 2Fc2)/3 (Δ/σ)max = 0.007
Δρmax = 0.22 e Å−3 Δρmin = −0.28 e Å−3
Special details
Experimental. Please note cell_measurement_ fields are not relevant to area detector data, the entire data set is used to refine the cell, which is indexed from all observed reflections in a 10 degree phi range.
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes.
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger.
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
x y z Uiso*/Ueq
O1 1.0233 (5) 0.56379 (17) 0.8951 (2) 0.0272 (6)
O2 0.8375 (5) 0.37968 (15) 0.6439 (2) 0.0277 (6)
O3 0.2063 (5) 0.29402 (17) 1.0335 (2) 0.0302 (6)
O4 0.3312 (5) 0.37320 (18) 1.2564 (2) 0.0287 (6)
H4 0.188 (8) 0.326 (3) 1.229 (3) 0.034*
N1 0.4571 (6) 0.4431 (2) 0.5244 (3) 0.0253 (7)
H1 0.452 (8) 0.409 (3) 0.469 (4) 0.030*
N2 0.8203 (7) 0.7503 (2) 0.4526 (3) 0.0301 (8)
H2 0.922 (8) 0.784 (3) 0.435 (3) 0.036*
C1 0.8558 (7) 0.4855 (2) 0.8585 (3) 0.0210 (8)
H1A 0.9837 0.4376 0.8422 0.025*
C2 0.6525 (7) 0.5072 (2) 0.7311 (3) 0.0202 (8)
H2A 0.4580 0.5169 0.7451 0.024*
C3 0.6552 (7) 0.4363 (2) 0.6300 (3) 0.0198 (7)
C4 0.2289 (7) 0.5057 (3) 0.4966 (3) 0.0249 (8)
H4A 0.2013 0.5319 0.5778 0.030*
H4B 0.0544 0.4744 0.4578 0.030*
C5 0.2770 (7) 0.5787 (2) 0.4074 (3) 0.0262 (9)
H5A 0.3016 0.5521 0.3259 0.031*
H5B 0.1063 0.6155 0.3883 0.031*
C6 0.5239 (7) 0.6368 (2) 0.4562 (3) 0.0242 (8)
C7 0.7173 (7) 0.6401 (2) 0.5761 (3) 0.0229 (8)
C8 0.7749 (7) 0.5910 (2) 0.6915 (3) 0.0183 (7)
C9 0.9907 (7) 0.6167 (2) 0.7875 (3) 0.0244 (8)
C10 1.1662 (7) 0.6868 (3) 0.7815 (3) 0.0284 (9)
H10 1.3128 0.7014 0.8517 0.034*
C11 1.1207 (7) 0.7352 (2) 0.6697 (4) 0.0288 (9)
C12 0.9010 (7) 0.7118 (2) 0.5717 (3) 0.0249 (8)
C13 0.5985 (8) 0.7047 (3) 0.3855 (3) 0.0297 (9)
H13 0.5065 0.7181 0.3005 0.036*
C14 0.7172 (7) 0.4572 (2) 0.9658 (3) 0.0211 (8)
C15 0.7808 (7) 0.4961 (2) 1.0836 (3) 0.0254 (8)
H15 0.9135 0.5424 1.0987 0.031*
C16 0.6509 (7) 0.4678 (3) 1.1822 (3) 0.0265 (8)
H16 0.6957 0.4948 1.2639 0.032*
C17 0.4596 (7) 0.4013 (2) 1.1600 (3) 0.0235 (8)
C18 0.3967 (7) 0.3605 (2) 1.0414 (3) 0.0224 (8)
C19 0.5267 (7) 0.3883 (2) 0.9439 (3) 0.0243 (8)
H19 0.4855 0.3603 0.8628 0.029*
C20 0.1393 (9) 0.2442 (3) 0.9195 (3) 0.0347 (9)
H20A 0.0760 0.2834 0.8468 0.042*
H20B −0.0104 0.2026 0.9258 0.042*
H20C 0.3059 0.2121 0.9072 0.042*
Atomic displacement parameters (Å2)
U11 U22 U33 U12 U13 U23
O1 0.0309 (14) 0.0320 (15) 0.0196 (13) −0.0107 (12) 0.0070 (11) −0.0003 (12)
O2 0.0350 (14) 0.0220 (14) 0.0271 (13) 0.0068 (13) 0.0092 (11) −0.0017 (11)
O3 0.0382 (15) 0.0291 (14) 0.0253 (13) −0.0074 (13) 0.0114 (12) −0.0014 (12)
O4 0.0320 (13) 0.0376 (16) 0.0189 (12) −0.0027 (13) 0.0110 (11) 0.0005 (12)
N1 0.0330 (17) 0.0222 (17) 0.0220 (18) −0.0033 (15) 0.0091 (15) −0.0054 (14)
N2 0.036 (2) 0.0203 (17) 0.0371 (19) −0.0026 (16) 0.0153 (16) 0.0080 (15)
C1 0.0222 (17) 0.0227 (19) 0.0179 (18) −0.0015 (16) 0.0035 (15) −0.0005 (14)
C2 0.0222 (17) 0.0197 (18) 0.0202 (18) −0.0012 (16) 0.0076 (15) −0.0019 (15)
C3 0.0221 (17) 0.0181 (18) 0.0209 (18) −0.0039 (17) 0.0084 (16) 0.0005 (15)
C4 0.0222 (18) 0.030 (2) 0.023 (2) −0.0012 (17) 0.0050 (15) 0.0009 (16)
C5 0.0280 (19) 0.032 (2) 0.0197 (18) 0.0056 (18) 0.0070 (16) 0.0012 (16)
C6 0.027 (2) 0.023 (2) 0.026 (2) 0.0059 (17) 0.0121 (16) 0.0009 (16)
C7 0.0262 (19) 0.018 (2) 0.028 (2) 0.0027 (16) 0.0127 (16) −0.0028 (16)
C8 0.0214 (16) 0.0162 (17) 0.0196 (17) 0.0018 (15) 0.0098 (15) −0.0007 (14)
C9 0.0263 (19) 0.028 (2) 0.0217 (18) −0.0019 (17) 0.0126 (16) −0.0013 (16)
C10 0.0293 (19) 0.031 (2) 0.027 (2) −0.0085 (19) 0.0107 (16) −0.0137 (18)
C11 0.031 (2) 0.023 (2) 0.039 (2) −0.0093 (18) 0.0210 (18) −0.0054 (18)
C12 0.031 (2) 0.017 (2) 0.029 (2) 0.0013 (17) 0.0124 (17) 0.0009 (16)
C13 0.036 (2) 0.029 (2) 0.0264 (19) 0.0074 (19) 0.0123 (17) 0.0090 (18)
C14 0.0222 (17) 0.0252 (19) 0.0161 (17) 0.0035 (16) 0.0048 (14) 0.0002 (15)
C15 0.0261 (19) 0.025 (2) 0.026 (2) 0.0004 (17) 0.0072 (16) 0.0007 (16)
C16 0.0280 (18) 0.031 (2) 0.0200 (18) 0.0011 (17) 0.0043 (15) −0.0030 (16)
C17 0.0254 (17) 0.030 (2) 0.0157 (18) 0.0050 (18) 0.0063 (15) 0.0055 (16)
C18 0.0227 (18) 0.0218 (18) 0.0227 (18) 0.0018 (16) 0.0051 (15) 0.0028 (16)
C19 0.0268 (18) 0.029 (2) 0.0177 (17) 0.0018 (18) 0.0066 (14) −0.0003 (16)
supporting information
sup-5
Acta Cryst. (2001). E57, o81–o82
Geometric parameters (Å, º)
O1—C9 1.389 (4) C5—C6 1.500 (5)
O1—C1 1.456 (4) C6—C13 1.376 (5)
O2—C3 1.227 (4) C6—C7 1.428 (5)
O3—C18 1.366 (4) C7—C12 1.422 (5)
O3—C20 1.417 (4) C7—C8 1.422 (5)
O4—C17 1.380 (4) C8—C9 1.367 (4)
N1—C3 1.331 (4) C9—C10 1.380 (5)
N1—C4 1.451 (5) C10—C11 1.384 (5)
N2—C13 1.360 (5) C11—C12 1.383 (5)
N2—C12 1.384 (4) C14—C15 1.370 (5)
C1—C14 1.509 (5) C14—C19 1.391 (5)
C1—C2 1.546 (4) C15—C16 1.404 (5)
C2—C8 1.510 (5) C16—C17 1.367 (5)
C2—C3 1.532 (5) C17—C18 1.389 (5)
C4—C5 1.516 (5) C18—C19 1.393 (4)
C9—O1—C1 107.0 (2) C7—C8—C2 132.2 (3)
C18—O3—C20 119.1 (3) C8—C9—C10 125.2 (3)
C3—N1—C4 128.1 (3) C8—C9—O1 113.2 (3)
C13—N2—C12 108.2 (3) C10—C9—O1 121.6 (3)
O1—C1—C14 110.2 (3) C9—C10—C11 117.6 (3)
O1—C1—C2 106.5 (3) C12—C11—C10 118.6 (3)
C14—C1—C2 115.2 (3) C11—C12—N2 128.1 (3)
C8—C2—C3 109.9 (2) C11—C12—C7 124.6 (3)
C8—C2—C1 102.1 (3) N2—C12—C7 107.2 (3)
C3—C2—C1 111.9 (3) N2—C13—C6 111.5 (3)
O2—C3—N1 122.6 (3) C15—C14—C19 120.0 (3)
O2—C3—C2 121.6 (3) C15—C14—C1 121.5 (3)
N1—C3—C2 115.8 (3) C19—C14—C1 118.5 (3)
N1—C4—C5 113.8 (3) C14—C15—C16 120.4 (3)
C6—C5—C4 115.6 (3) C17—C16—C15 119.6 (3)
C13—C6—C7 105.5 (3) C16—C17—O4 119.9 (3)
C13—C6—C5 122.6 (3) C16—C17—C18 120.5 (3)
C7—C6—C5 131.9 (3) O4—C17—C18 119.6 (3)
C12—C7—C8 114.9 (3) O3—C18—C17 114.2 (3)
C12—C7—C6 107.5 (3) O3—C18—C19 125.9 (3)
C8—C7—C6 137.5 (3) C17—C18—C19 119.8 (3)
C9—C8—C7 119.0 (3) C14—C19—C18 119.7 (3)
C9—C8—C2 108.7 (3)
C9—O1—C1—C14 −141.6 (3) C8—C9—C10—C11 0.0 (5)
C9—O1—C1—C2 −16.1 (3) O1—C9—C10—C11 179.1 (3)
O1—C1—C2—C8 13.1 (3) C9—C10—C11—C12 1.1 (5)
C14—C1—C2—C8 135.5 (3) C10—C11—C12—N2 −179.4 (3)
O1—C1—C2—C3 130.6 (3) C10—C11—C12—C7 −0.9 (5)
C4—N1—C3—O2 179.1 (3) C13—N2—C12—C7 −0.3 (4)
C4—N1—C3—C2 −4.2 (4) C8—C7—C12—C11 −0.4 (5)
C8—C2—C3—O2 99.8 (4) C6—C7—C12—C11 −178.8 (3)
C1—C2—C3—O2 −12.9 (4) C8—C7—C12—N2 178.3 (3)
C8—C2—C3—N1 −76.9 (3) C6—C7—C12—N2 −0.1 (4)
C1—C2—C3—N1 170.4 (3) C12—N2—C13—C6 0.6 (4)
C3—N1—C4—C5 103.1 (4) C7—C6—C13—N2 −0.7 (4)
N1—C4—C5—C6 −62.5 (4) C5—C6—C13—N2 177.0 (3)
C4—C5—C6—C13 178.7 (3) O1—C1—C14—C15 −7.8 (5)
C4—C5—C6—C7 −4.4 (5) C2—C1—C14—C15 −128.3 (4)
C13—C6—C7—C12 0.4 (4) O1—C1—C14—C19 173.9 (3)
C5—C6—C7—C12 −176.9 (3) C2—C1—C14—C19 53.5 (4)
C13—C6—C7—C8 −177.4 (4) C19—C14—C15—C16 −1.1 (5)
C5—C6—C7—C8 5.2 (7) C1—C14—C15—C16 −179.4 (3)
C12—C7—C8—C9 1.5 (4) C14—C15—C16—C17 −0.1 (5)
C6—C7—C8—C9 179.2 (4) C15—C16—C17—O4 180.0 (3)
C12—C7—C8—C2 −173.6 (3) C15—C16—C17—C18 1.1 (5)
C6—C7—C8—C2 4.1 (6) C20—O3—C18—C17 −176.1 (3)
C3—C2—C8—C9 −124.7 (3) C20—O3—C18—C19 3.4 (5)
C1—C2—C8—C9 −5.7 (3) C16—C17—C18—O3 178.8 (3)
C3—C2—C8—C7 50.8 (5) O4—C17—C18—O3 −0.1 (5)
C1—C2—C8—C7 169.8 (3) C16—C17—C18—C19 −0.8 (5)
C7—C8—C9—C10 −1.4 (5) O4—C17—C18—C19 −179.7 (3)
C2—C8—C9—C10 174.8 (3) C15—C14—C19—C18 1.4 (5)
C7—C8—C9—O1 179.5 (3) C1—C14—C19—C18 179.7 (3)
C2—C8—C9—O1 −4.3 (4) O3—C18—C19—C14 180.0 (3)
C1—O1—C9—C8 13.1 (4) C17—C18—C19—C14 −0.5 (5)
C1—O1—C9—C10 −166.0 (3)
Hydrogen-bond geometry (Å, º)
D—H···A D—H H···A D···A D—H···A
N1—H1···O4i 0.79 (4) 2.29 (4) 3.002 (4) 150 (4)
N2—H2···O2ii 0.76 (4) 2.15 (4) 2.909 (4) 171 (3)
O4—H4···O3 1.00 (4) 2.17 (3) 2.631 (5) 106 (2)
C1—H1A···O2 1.00 2.27 2.794 (4) 111
C11—H11···O4iii 0.95 2.51 3.367 (4) 150
C15—H15···O1 0.94 2.37 2.740 (4) 103