Estimating the Total Number of Alleles Using a Sample Coverage Method
Shu-Pang Huang and B. S. Weir
Program in Statistical Genetics, Department of Statistics, North Carolina State University, Raleigh, North Carolina 27695-7566 Manuscript received September 29, 2000
Accepted for publication August 31, 2001
ABSTRACT
Previously reported methods for estimating the number of different alleles at a single locus in a population have not described a useful general result. Using the number of alleles observed in a sample gives an underestimate for the true number of alleles. The similar problem of estimating the number of species in a population was first investigated in 1943. In this article we use the sample coverage method proposed by Chao and Lee in 1992 to estimate the number of alleles in a population when there are unequal allele frequencies. Simulation studies under the recurrent mutation model show that, for reasonable sample sizes, a significantly better estimate of the true number can be obtained than that using only the observed alleles. Results under the stepwise mutation model and infinite-allele model are presented. Possible applications include improving the characterization of the prior distribution for the allele frequencies, adjusting the estimates of genetic diversity, and estimating the range of microsatellite alleles.
A
major topic in population genetics is the character- Bayesian philosophies (BungeandFitzpatrick1993). ization of the distribution of allele frequencies for It is quite straightforward to relate our problem to theirs a population. Some theoretical results under different if we treat allele types as species.evolutionary forces have been proposed (Crow and Most of the estimating methods are based on sampling Kimura1970). For example, under the recurrent muta- theory. If the underlying population is finite, it is natural tion model (RMM), which assumes that every mutation to use the hypergeometric model. When the population will lead only to another preexisting allele, the stationary size is large, however, the multinomial model is a good distribution for allele frequencies will be the Dirichlet approximate model to use. Several methods have been distribution (Griffiths1979). This model assumes that proposed under the multinomial model (Bunge and the number of allelesM is known. Unfortunately, we Fitzpatrick1993). We use the sample coverage (SC) do not know this number in general. Many other popu- method proposed byChaoandLee(1992). It is a non-lation genetic parameters are also associated with allele parametric method and its performance is better than numbers. For instance, the genetic diversity (Weir other methods (Bungeet al.1995).
1996), defined as 1⫺ RM
i⫽1p2i, where piis the frequency We describe how to obtain estimators based on the for theith allele, faces the same problem. The parame- sample coverage method, which is followed by simula-terMis usually estimated by the number of alleles ob- tion studies using the coalescent process under different served in a sample. This will, of course, underestimate mutation models. Examples are given to illustrate
appli-the true allele number. cations of this method.
The same problem has been recognized for a long time by ecologists who want to use a sample to estimate
the number of species (or individuals) in a population. METHOD
Since Fisher et al. (1943) first proposed a statistical
Suppose there are M different alleles for a locus in model to estimate the number of species in a
popula-a populpopula-ation. A rpopula-andom spopula-ample of n alleles is drawn tion, it has been an active research field with
applica-from the population. LetXi be the number of the ith tions in many other fields. For example,Lewontinand
type of allele observed in the sample andDthe number Prout(1956) derived a maximum-likelihood estimator
of different observed allele types. Furthermore, letfjbe under the assumption of equal frequencies and applied
the number of alleles that havejrepresentatives in the it to estimate the number of genes on a chromosome.
sample. It is easy to see that D⫽Rn
j⫽1fj and n⫽ RMi⫽1 Several methods have been proposed to manage the
Xi⫽ Rnj⫽1j fj. If theith allele type has frequencypiin the unequal frequencies situation, including both
paramet-population, then under the equal frequencies assump-ric and nonparametassump-ric approaches under frequentist or
tion (i.e., p1⫽ p2 ⫽ · · ·⫽ pM ⫽1/M), the likelihood for the given sample is
Corresponding author:B. S. Weir, Bioinformatics Research Ctr.,
Cam-pus Box 7566, North Carolina State University, Raleigh, NC 27695- L(M)⫽
冤
n!兿
Di⫽1Xi!
兿
nj⫽1fj!冥 冤
M! (M⫺D)!冢
1
M
冣
n冥
. 7566. E-mail: [email protected]The maximum-likelihood estimator Mˆ for M can be Instead of estimating the number of classes M di-rectly, if we can estimate the percentage (denoted by derived as
C) of classes that are represented in the sample, the quantityD/C, the ratio of the observed classes and their
n
Mˆ ⫽
兺
Mˆ
j⫽Mˆ⫺D⫹1 1
j ≈ln
冢
MˆMˆ ⫺ D⫹ 1
冣
(1) total percentage, can serve as an estimate for theparam-eterM.A formal definition for the parameterC, namely, (Feller1950).
sample coverage, is On the other hand, the probability mass function of
the numberUof unseen allele types is
C⫽
兺
M
i⫽1
piI[Xi⬎ 0].
PU ⫽
冢
M
U
冣
兺
M⫺U
⫽0
(⫺1)
冢
M⫺ U
冣冢
1⫺U⫹
M
冣
n
This is the sum of the probabilities of classes observed in a sample. Now if all allele types have the same
fre-≈ e⫺ U
U! quency in the population,i.e.,p1 ⫽ p2⫽ · · ·⫽ pM ⫽
1/M, then (Feller1950), where ⫽Me⫺n/M. This distribution
con-C⫽
兺
M
i⫽1 1
MI(Xi⬎ 0)⫽ D M
verges to a Poisson () distribution asnincreases. But, since at least one type will appear in the sample, the appropriate distribution forUis actually the truncated
M⫽D
C.
distribution of PU,i.e.,
If we can estimate the sample coverageC, the estimate
f(U)⫽ P(U)
1⫺ P(U⫽M). ofMwill follow directly. The quantity Chas been well
studied.Good(1953) andEsty(1982, 1986) used the Taking the expectation ofU, and by suitable
transforma-following estimator proposed by Turing (Good 1953): tion, we have
Cˆ ⫽1⫺f1/n.
E(U)≈
冤
兺
M⫺2Z⫽0
e⫺ Z
Z!
1
1⫺P(z⫽ M⫺1)
冥
Under the equal probability case, we haveMˆ1⫽D/Cˆ (3)
(letz ⫽U⫺1)
(DarrochandRatcliff1958). Compared to the
maxi-≈(because
兺
M⫺2 Z⫽0e⫺Z
Z!⫽1⫺P(z⫽ M⫺1)) mum-likelihood estimator (MLE) under the equiproba-ble population, this estimator is very efficient. Both
⫽Me⫺n/M
. estimators, however, suffer the same problem of
under-estimating M when the pi’s are not all equal. But in
BecauseU⫽M⫺D, we have the definition ofC, we did not require allp
i’s to be the same. Chao and Lee (1992) therefore proposed the
E(D) ≈M(1 ⫺e⫺n/M),
following approach to obtain an adjusted estimator for
or M.They used a Taylor series to expandE(D)/E(C) up
to the second order with respect to the equal probability
n M⫽ln
冢
M
M⫺ E(D)
冣
. (2) pointp1⫽ p2⫽· · ·⫽ pM⫽1/M.This providesFrom Equations 1 and 2, the approximate maximum- E(D)
E(C)≈M⫺
n(1⫺ p)n⫺1
E(C) ␥
2, (4)
likelihood estimateMˆ ofMsatisfies
where␥ ⫽[Ri(pi ⫺p)2/M]1/2/pis the coefficient of vari-D⫽Mˆ[1⫺ e⫺n/Mˆ
]
ation (CV) and is alwaysⱖ0. By observing that (LewontinandProut1956).Harris(1968) obtained
E(f1)⫽
兺
inpi(1⫺pi)n⫺1 the asymptotic variance for this estimator
Var(Mˆ)⫽M/[en/M⫺(n/M)⫺1]. ≈
n(1⫺p)n⫺1⫺ [2E(f
2)⫺ 3E(f3)]␥2, (5)
The assumption of equiprobable frequencies is usually we can substitute Equation 5 into Equation 4 and get unrealistic and Mˆ will therefore underestimate the the estimating function
number of allele types. To solve this problem, several
works have proposed different distributions to model M≈ E(D)
E(C)⫹
E(f1)
E(C)␥
2. (6)
the so-called “capture” probability for each class (e.g.,
species, allele types;Engen1978). Although those para- GoodandToulmin(1956) obtained the equation metric approaches can deal with the heterogeneous
problem in some way, they are still highly dependent ␥2⫽ M
兺
n
j j(j⫺1)E(fj)
By substituting Equation 3 andfj into the formula, we est is the actual number of alleles in the simulated popu-lation rather than the number of all possible alleles. can get an estimate for␥2:
The simulation consists of two levels. In the first level, we simulate a population of size 10,000 and calculate
␥ˆ2⫽max
Mˆ1
兺
n
j j(j⫺ 1)fj [n(n ⫺1)] ⫺1, 0
. (7) allele types and frequencies on the basis of the mutation
model. For the second level, we randomly choose a Replacing the expected quantities by observed values
sample of size 100 from the simulated population and and combining with Equation 7 leads to an estimate of
use our method to estimate the number of alleles in
M:
that particular population. We take 400 samples from each of 400 simulated populations.
Mˆ2⫽
D Cˆ ⫹
n(1⫺Cˆ)
Cˆ ␥ˆ
2. (8)
The simulation algorithm for the first level is based on the coalescent process (Kingman 1982). Hudson The bias of␥ˆ2 is greater when ␥ is large. An adjusted
(1993) gave a general description of simulation meth-estimator,␥˜2, of␥2is recommended byChaoandLee
ods. The results for each simulated population are sum-(1992),
marized as
Mˆ3⫽
D Cˆ ⫹
n(1⫺Cˆ)
Cˆ ␥˜
2, (9)
Bias⫽
兺
400k⫽1Mˆki ⫺Msimi
400 ,
where
Standard error⫽
冪
兺
400k⫽1 (Mˆki ⫺ Mi)2
400 ,
␥˜2⫽ ␥ˆ2
冤
1⫹n(1 ⫺Cˆ)兺
j(j⫺1)fj n(n⫺ 1)Cˆ冥
.with Mi⫽
兺
400 k⫽1Mˆki400 For the variance of the estimators, recall that all of
the quantities used in the estimators are functions of
Est. no. of the multinomial counts (f0,f1, · · · ,fn). Note that under
this setting, the sample sizen⫽ Rn
j⫽1jfjis also a random
unobserved alleles⫽
兺
400k⫽1(Mˆki ⫺ Dki)
400 , (11)
variable. The asymptotic variance for the estimator can be derived using standard asymptotic approach,
where Mˆk
i and Dki are the estimate and the observed number of alleles for thekth sample from theith simu-Vaˆr(Mˆi)≈
兺
n
j⫽1
兺
nk⫽1 Mˆi
fj
Mˆi
fk
cov(fj,fk), (10)
lated population andMˆsim
i is the actual number of alleles in theith simulated population. For each population, where
we plot these three quantities against the number of alleles in that population. Note that, because of the evolutionary process, the CV among allele frequencies cov(fj,fk)⫽
fj
冢
1⫺ fjMˆi冣 ifj⫽k ⫺fjfk
Mˆi
ifj⬆ k. is usually large. Since the first two estimatorsM
ˆ1 and Mˆ2usually underestimate the trueMwhen CV is large (ChaoandLee1992), onlyMˆ3is used in the simulation studies to measure the performance of the sample cover-age method.
SIMULATION STUDY Simulation results under RMM: Since the magnitude
of CV is reported to be an important effect on perfor-Simulation studies were performed under the
follow-mance, particular configurations ofpi’s are selected to ing three mutation models:
reflect different CV levels. We chose Zipf’s law to specify 1. RMM: Every mutation produces a preexisting type allele frequencies because “Zipf’s laws are probability of allele. It is a reversible process. There is no restric- distributions on the positive integers which decay alge-tion for an allele to mutate to another type as long braically. Such laws have been shown empirically to de-as the mutation rate toward a particular type is not scribe . . . the distribution of biological genera and
zero. species” (Chen1980). Its form isp
iⵑci⫺(1⫹␣)asi→∞. 2. Infinite-allele model (IAM): Each mutation creates We select␣ ⫽ 0 so thatp
i⫽ci⫺1.
a new allele type. Under this particular model, suppose the rate for all
3. Stepwise mutation model (SMM): Each mutation is other allele types mutating to a particular type i isi. more likely to change to its adjacent type(s). When the population reaches equilibrium, the allele frequency for alleleiisi/., where.⫽ RMi⫽1i. In our Note that, even under the RMM where the number
simulation, we chosei ⫽0.0001b/(b ⫹ i) so that the of alleles is set up beforehand, the actual number of
slightly adjusted Zipf’s law is alleles in a population is unknown because of the
evolu-tionary process. For each simulated population, we may
pi⫽ i/.⫽
冤
兺
Mj⫽1
(b⫹i)/(b⫹j)
冥
⫺1
⫽c(b⫹i)⫺1. (12)
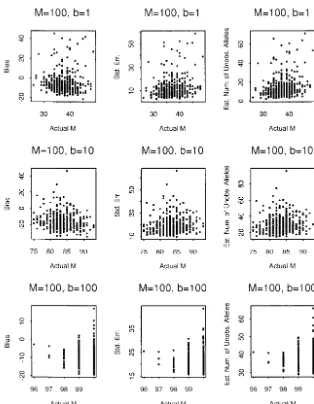
inter-Figure 1.—The simulation study for the RMM whenM⫽20. Thex-axes are the actual numbers of alleles obtained from the simu-lated populations and they-axes are the quanti-ties described in Equation 11.
The role ofbhere is to adjust the CV of allele
frequen-pi→j⫽
␣P(1 ⫺P)j⫺i⫺1 ifj⬎i
(1⫺ ␣)P(1 ⫺P)i⫺j⫺1 ifj⬍i, cies. The larger the b, the smaller the CV. We choseb
to be 1, 10, and 100 to represent high, medium, and
i.e., the transition probability from typei to typej de-low CV values, respectively.
pends on the absolute value of the size difference (|i⫺
From Figures 1 and 2, we observe that the biases of
j|). If only the size change is concerned, the distribution estimates are all close to 0 when M is small but the
of size change is geometrically distributed. Since estimates exhibit some negative bias when M is large.
This also shows that the number of unobserved alleles
兺
∞j⬎i
pi→j⫽ ␣, could be very large. We also note that, when CV is large,
the standard error of the estimator is also large.
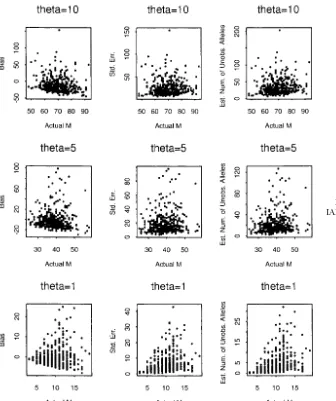
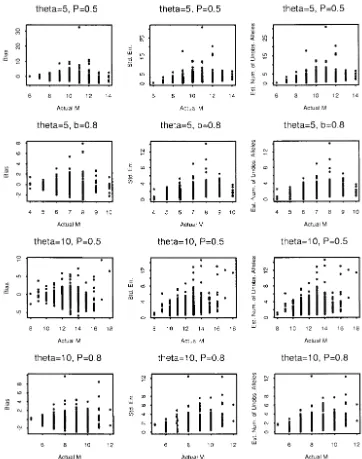
Simulation results under IAM: In the infinite-allele the parameter ␣ is the probability of increasing size case, the parameter ⫽4Nis selected to be {10, 5, 1}. when a mutation occurs. Hence, if␣ ⬎ 0.5, mutations The results are shown in Figure 3. From the third col- tend to increase the size. The parameter Phas the ef-umn of Figure 3, we see that the difference between fect of determining the magnitude of size change. The the observed and estimated number of alleles increases size change reaches its maximum when P ⫽ 0.5 and asvalues increase. This is reasonable since, when the becomes smaller whenPapproaches its boundaries (0 mutation rate increases, more distinct alleles are ex- or 1).
Although there is no restriction on the size change pected in the population under the IAM.
Simulation results under SMM:Under the SMM, we under the model, observations show that each mutation is more likely to change an allele to an adjacent allele. adopted the model proposed byFuandChakraborty
Figure 2.—The simulation study for the RMM whenM⫽100.
when the transition probability greater than that step data from nine populations of honey bee (Apis Mellifera L.) subspecies. The adequacy of two mutation models is less than some critical valueε. The maximum steps
is determined by usually used for microsatellite, IAM and RMM, are tested
by comparing the observed and theoretical number of alleles. The authors claimed that the IAM fits the data
s⫽1⫺ ⫺log10(ε)⫹log10(max{␣, 1⫺ ␣})⫹ log10(P)
log10(1⫺ P) . better and argued that the reason may be because the
majority of the microsatellites they used in the study In our simulation study, we set␣ ⫽0.5 and chose two
contain repeats of several different length motifs. They
Pvalues⫽{0.5, 0.8} and two values⫽{5, 10}. Results
also provided estimates of effective population size are shown in Figure 4. Theεvalue was set to 0.001, so
(2Ne) and mutation rates () for each locus on the basis that the maximum steps forP ⫽0.5 and 0.8 are eight
of some assumptions. We chose the subspeciesTiznitas and four, respectively. Simulation results for different
an example to demonstrate the use of our method. The
␣values are very similar to Figure 4 and are not shown
allele frequencies in Estoup et al.’s data are changed here. On the basis of Figure 4, we observe that, even
to allele counts to fit our need. The data are listed in though we setP⫽ 0.5 and each mutation can change
Table 1. an allele by up to 8 units, the number of alleles observed
Under the IAM, Crow andKimura (1970) derived in the sample is still very limited. Under the same
muta-the expected number of alleles that can be maintained tion rate and sample size, we can observe many more
in a population,MCK, to be alleles under IAM than SMM. As a result, the estimates
are not very different from the observed numbers.
MCK⫽
冮
11/2Ne
(1⫺ x)⫺1x⫺1dx, (13)
EXAMPLES AND APPLICATIONS
Figure 3.—The simulation study for the IAM.
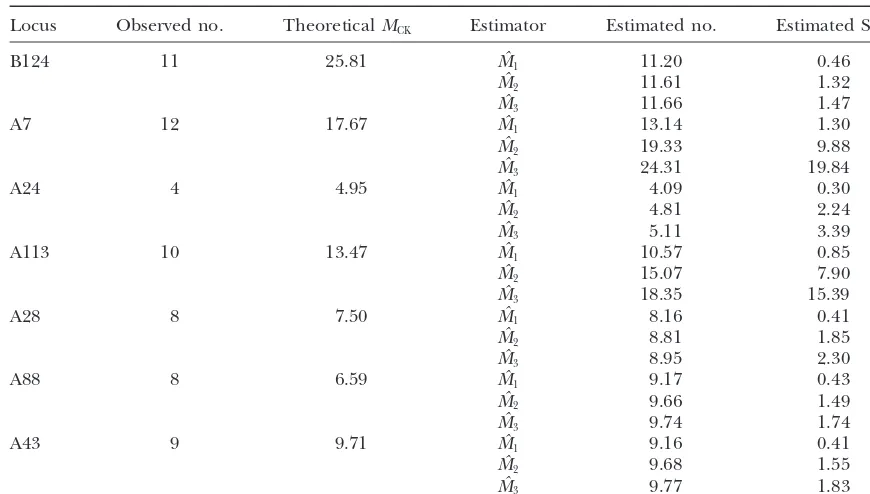
Table 1 together with Equation 13 we can get the theo- relatively small and all the alleles in the data are different (i.e., D ⫽ f1 ⫽ n), we have cˆ⫽ 0 and hence Mˆ ⫽ ∞. retical number of alleles for the population. Table 2
shows the theoretical allele numbers and our three esti- Although in this case we would expect there are many alleles in the population, this estimate is not useful. For mates. The estimated allele numbers are generally close
toMCK. this situation,Chao(1984) derived an estimateMˆlow⫽
D ⫹ f1(f1 ⫺ 1)/2 for the lower bound of M. We use Note that, in some cases, MCK is smaller than the
observed number. This reflects the problem that the this estimate when simulations would give an infinite estimate. In real data, fortunately, this kind of situation two parameters, effective population size and mutation
rate, are generally difficult to estimate and have high is almost impossible.
We also note that when the CV is large, the estimates uncertainty. One advantage of the sample coverage
method is that we do not need to estimate the effective still have a large negative bias. One way to conquer this problem is to use the stabilization technique (Haasand population size and mutation rate before estimating the
allele numbers. Stokes1998) that uses only those alleles appearing less
thanktimes in the sample to obtain the estimates and Besides the direct applications in improving the
esti-mation of gene diversity and characterization of prior then adding alleles having more thankrepresentatives. For example, suppose we haveralleles appearing more distribution of allele frequencies, under SMM, we can
use this information to predict the range of allele sizes thanktimes for a total ofnralleles in the sample. We first use the remaining data (n ⫺ nr) to estimate the as well.
allele number (M⫺r) and then add theralleles back.
Our estimates, therefore, will be Mˆ ⫽ ⫹ r.
DISCUSSION
Since the alleles with high frequencies are not used in estimation, the CV will be reduced significantly. The A drawback of the sample coverage method is that,
Figure4.—The simulation study for the SMM.
removed from the estimation process is that, since they k ⫽ 10 works well (data not shown). We should be careful, however, in applying this technique to real ge-are so abundant, observing them will not provide any
useful information about unseen alleles and hence they netic data, because the variation of frequencies among alleles could be very large. For small samples, removing are not helpful in estimating the total number. The
choice ofkis somewhat arbitrary. Simulations show that alleles with ⬎10 copies from the data may result in an
TABLE 1
The data ofEstoupet al.(1995)
Locus Mutation rate n Allele counts
B124 1.002⫾0.138⫻10⫺3 56 f
1⫽1,f2⫽2,f3⫽2,f4⫽1,f5⫽2,f6⫽1,f12⫽1,f13⫽1
A7 0.686⫾0.138⫻10⫺3 46 f
1⫽4,f2⫽3,f3⫽1,f4⫽1,f5⫽1,f6⫽1,f18⫽1
A24 0.192⫾0.042⫻10⫺3 48 f
1⫽1,f6⫽1,f13⫽1,f28⫽1
A113 0.523⫾0.084⫻10⫺3 56 f
1⫽3,f2⫽3,f5⫽1,f7⫽1,f12⫽1,f23⫽1
A28 0.291⫾0.056⫻10⫺3 52 f
1⫽1,f2⫽2,f3⫽1,f6⫽1,f8⫽1,f12⫽1,f18⫽1
A88 0.256⫾0.051⫻10⫺3 54 f
1⫽1,f2⫽2,f3⫽1,f4⫽1,f6⫽1,f9⫽1,f13⫽1,f14⫽1
A43 0.377⫾0.067⫻10⫺3 58 f
TABLE 2
Results for the data ofEstoupet al.(1995)
Locus Observed no. TheoreticalMCK Estimator Estimated no. Estimated SE
B124 11 25.81 Mˆ1 11.20 0.46
Mˆ2 11.61 1.32
Mˆ3 11.66 1.47
A7 12 17.67 Mˆ1 13.14 1.30
Mˆ2 19.33 9.88
Mˆ3 24.31 19.84
A24 4 4.95 Mˆ1 4.09 0.30
Mˆ2 4.81 2.24
Mˆ3 5.11 3.39
A113 10 13.47 Mˆ1 10.57 0.85
Mˆ2 15.07 7.90
Mˆ3 18.35 15.39
A28 8 7.50 Mˆ1 8.16 0.41
Mˆ2 8.81 1.85
Mˆ3 8.95 2.30
A88 8 6.59 Mˆ1 9.17 0.43
Mˆ2 9.66 1.49
Mˆ3 9.74 1.74
A43 9 9.71 Mˆ1 9.16 0.41
Mˆ2 9.68 1.55
Mˆ3 9.77 1.83
MCKassumesNe⫽1617.
unrealistically large estimate because only a very small esting to know the allele numbers for a whole species or how many alleles are shared between subspecies. amount of data is used in the estimation. Hence, we
did not use this technique in Table 2. Chaoet al. (2000) extended the method to this kind
of multipopulation problem in ecology. Further studies Besides these technical problems, some concerns still
need to be addressed. For example, what should we do are needed to adjust their method to population ge-netics.
when some alleles have been reported previously but are
not in the present sample? If there are several samples The authors thank anonymous reviewers for their help. This work available, should we combine them to give a larger sam- was supported in part by National Institutes of Health grant GM 45344. ple or should we use them separately? For the first
prob-lem, assuming there areLalleles we know exist but are
not in our sample, possible solutions are LITERATURE CITED
Bunge, J.,and M. Fitzpatrick, 1993 Estimating the number of 1. Mˆ⬘ ⫽Mˆ ⫹L, or species: a review. J. Am. Stat. Assoc.88:364–373.
Bunge, J., M. FitzpatrickandJ. Handley,1995 Comparison of 3 2. replacenbyn⬘ ⫽n⫹L,D⬘ ⫽D⫹L, andf⬘1 ⫽f1⫹
estimators of the number of species. J. Appl. Stat.22:45–59.
Lto obtainMˆ⬘, or
Chao, A.,1984 Nonparametric estimation of the number of the 3. sinceMˆ has already estimated the unseen alleles from classes in a population. Scand. J. Stat.11:265–270.
Chao, A.,andS.-M. Lee,1992 Estimating the number of classes via the data, it should already include thoseL alleles.
sample coverage. J. Am. Stat. Assoc.87:210–217. We need only adjust our estimates whenMˆ ⬍ D⫹
Chao, A.,andP. Tsay,1998 A sample coverage approach to
multi-Land set Mˆ⬘ ⫽D⫹L. ple-system estimation with application to census undercount. J.
Am. Stat. Assoc.93:283–293.
For the second problem, besides the method of com- Chao, A., W. H. Hwang, Y. C. ChenandC. Y. Kuo,2000 Estimating the number of shared species in two communities. Stat. Sinica bining data, if the samples are from contemporary
gen-10:227–246.
erations, we can regard them all as a sample from a Chen, W.-C.,1980 On the weak form of Zipf’s law. J. Appl. Probab. capture-recapture experiment. We may use any of sev- 17:611–622.
Crow, J., andM. Kimura,1970 Introduction to Population Genetics eral methods (e.g., Lee and Chao 1994; Norris and
Theory.Harper & Row, New York.
Pollock 1996) for capture-recapture data. Recently Darroch, J. N.,and D. Ratcliff, 1958 A note on capture and ChaoandTsay(1998) proposed a method to estimate recapture estimation. Biometrics36:149–153.
Engen, S., 1978 Stochastic Abundance Model. Chapman & Hall,
M under a multisystem sample scheme that may offer
London.
inter-hierarchical genetic structure and test of the infinite allele and Haas, P.,andL. Stokes,1998 Estimating the number of classes in a finite population. J. Am. Stat. Assoc.93:1475–1487.
stepwise mutation models. Genetics140:679–695.
Esty, W. W.,1982 Confidence intervals for the coverage of low Harris, B.,1968 Statistical inference in the classical occupancy prob-lem: unbiased estimation of the number of classes. J. Am. Stat. coverage samples. Ann. Stat.11:905–912.
Esty, W. W.,1986 The efficiency of Good’s nonparametric coverage Assoc.63:837–847.
Hudson, R. R.,1993 The how and why of generating gene genealo-estimator. Ann. Stat.14:1257–1260.
Feller, W.,1950 An Introduction to Probability Theory and Its Applica- gies, pp. 23–36 inMechanisms of Molecular Evolution, edited byN. TakahataandA. G. Clark.Sinauer Associates, Sunderland, MA. tions.John Wiley and Sons, New York.
Fisher, R., A. CorbetandC. Williams,1943 The relation between Kingman, J. F. C.,1982 The coalescent. Stoch. Proc. Appl.13:235–248.
Lee, S.,andA. Chao,1994 Estimating population-size via sample the number of species and the number of individuals in a random
sample of an animal population. J. Anim. Ecol.12:1257–1260. coverage for closed capture-recapture models. Biometrics50(1):
Fu, Y.-X.,andR. Chakraborty,1998 Simultaneous estimation of 88–97.
all the parameters of a stepwise mutation model. Genetics150: Lewontin, R.,andT. Prout,1956 Estimation of the number of
487–497. different classes in a population. Biometrics12:211–223.
Good, I. J.,1953 On the population frequencies of species and the Norris, J.,andK. Pollock,1996 Nonparametric mle under two estimation of population parameters. Biometrika40:237–264. closed capture recapture models with heterogeneity. Biometrics
Good, I. J.,andG. Toulmin,1956 The number of new species and 52(2): 639–649.
the increase of population coverage when a sample is increased. Weir, B. S.,1996 Genetic Data Analysis II.Sinauer Associates,
Sunder-Biometrika43:45–63. land, MA.
Griffiths, R. C.,1979 A transition density expansion for a