ABSTRACT
PETERSON, GEOFFREY COLIN LEE. Mean-Dependent Spatial Prediction Methods with Applications to Materials Sciences. (Under the direction of Dr. Brian Reich and Dr. Joseph Guinness.)
In this work, we explore spatial statistical models and their potential uses in materials
science, with particular emphasis on mean-dependent covariance structures. Materials science, which is the study and design of new solid materials, is a field ripe with complex data that
requires sophisticated statistical analysis. We investigate two important materials science data
applications: molecular dynamics simulations and crystallography diffraction analysis.
For the molecular dynamics simulations, computer simulations of the three-dimensional
atomic structures of materials explore how defects arise under simulated stresses. To overcome
the extreme computational burden of these simulations, we develop a spatial statistical model that emulates the final output of these computationally-intensive simulations. In a comparison
of multiple spatial regression methods, we find that principal component analysis best predicts
defects in the atomic structure.
For the crystallography analysis, radiation diffraction patterns are used to identify the
crys-tal lattice properties of a solid material. We investigate a statistical method of fusing the
multiple diffraction data sources to estimate the crystallographic parameters. We determine that variable weighting of the data sources significantly improves the accuracy of the estimates,
particularly when parts of the diffraction sources were unreliable due to data corruption.
Finally, as an extension of methods used in the materials sciences analyses, we develop a nonstationary spatial model where the covariance structure is indexed by the mean. Through
a simulation study, we explore the inferential and predictive capabilities of the model, showing
that it significantly improves upon a stationary spatial model under various circumstances. We then demonstrate these methods on daily precipitation data in Puerto Rico to show how they
© Copyright 2016 by Geoffrey Colin Lee Peterson
Mean-Dependent Spatial Prediction Methods with Applications to Materials Sciences
by
Geoffrey Colin Lee Peterson
A dissertation submitted to the Graduate Faculty of North Carolina State University
in partial fulfillment of the requirements for the Degree of
Doctor of Philosophy
Statistics
Raleigh, North Carolina
2016
APPROVED BY:
Dr. Donald Brenner Dr. Peter Bloomfield
Dr. Brian Reich
Co-chair of Advisory Committee
ACKNOWLEDGEMENTS
First and foremost, I would like to thank Dr. Brian Reich and Dr. Joseph Guinness as my advisors. Your guidance, support, and vast knowledge have helped brought this thesis to fruition.
Next, I would like to acknowledge our many collaborators from North Carolina State Uni-versity. Thank you to Dr. Don Brenner and Dong Li from the Department of Materials Sciences,
who generated the molecular dynamics simulation data. Thank you to Dr. Chris Fancher and
Dr. Jacob Jones from the Department of Materials Sciences and Engineering, who advised us on the crystallography analysis. Thank you to Dr. Adam Terando from the Department of Ecology,
who advised on the Puerto Rico precipitation analysis.
I would also like to acknowledge some of the many friends who have provided me with extensive personal support. In no particular order, thank you to Victoria Weber, Benjamin
Burner, Christropher Krut, Keith Shusterman, Timothy Scheisswohl, Kevin Garofalo and The
Raleigh Social Club, the friendly staff of Cup-A-Joes, and everyone else who made me laugh and smile along the way. And thank you to Adrienne Mazzo, who has made my life so bright,
beautiful, and full of joy.
Of course, I would not be anywhere without the love and support of my family. Thank you to my oldest brother Luc, for being a fantastic role model. Thank you to my brother Carl, for
being a constant source of diversion. Thank you to Annika, my dear sister and fellow
brown-eyed Lee, for being one of my best friends in the world. Thank you to my awesome sister-in-law Meghan and my beautiful niece Madeline, for growing the amount of love in my family. Thank
you to my grandmother Shirley, my aunts DeeAnn, Lori, Ann, and Pat, my uncles Bob, Brian,
and Drew, and my cousins Josh, Erin, Chris, Nick, and Drew.
Finally, thank you to my parents, Lee and Mary. You have given me everything in this
world, and I would not have been able to accomplish anything without your love and support.
I could not be more happy and proud to be your son.
DEDICATION
To the loving memories of my grandparents, Jay Peterson and Daisy Lee Shores.
TABLE OF CONTENTS
LIST OF TABLES . . . vi
LIST OF FIGURES . . . vii
Chapter 1 Spatial prediction of crystalline defects observed in molecular dy-namic simulations of plastic damage . . . 1
1.1 Introduction . . . 1
1.2 Data simulation and processing . . . 4
1.2.1 Simulation generation . . . 4
1.2.2 Quantifying materials defects . . . 5
1.2.3 Coarse graining . . . 6
1.3 Model description . . . 6
1.3.1 Prior Distributions . . . 7
1.3.2 Spatial Projections . . . 8
1.3.3 Basis Functions of Input Space . . . 12
1.4 Computing Details . . . 13
1.4.1 Reducing dimension to improve computation time . . . 14
1.4.2 Prediction Distribution . . . 15
1.5 Results . . . 15
1.6 Conclusion . . . 23
Chapter 2 Bayesian estimation of crystallographic parameters through weighted fusion of radiation diffraction data . . . 24
2.1 Introduction . . . 24
2.2 Statistical Model . . . 26
2.3 Computational Details for Parameter Estimation . . . 28
2.3.1 Background and Precision Parameters . . . 28
2.3.2 Crystallographic Parameters . . . 29
2.4 Simulation Study . . . 29
2.4.1 Data Generation . . . 29
2.4.2 Results and Discussion . . . 32
2.5 Conclusion . . . 42
Chapter 3 Mean-dependent nonstationary spatial models . . . 43
3.1 Introduction . . . 43
3.2 Nonstationary spatial model from conditionally stationary regimes . . . 45
3.3 Computation . . . 46
3.3.1 The Stationary Model . . . 47
3.3.2 The Nonstationary Model . . . 47
3.4 Evaluation of models . . . 47
3.4.1 Test for Nonstationarity . . . 48
3.4.2 Prediction Distribution . . . 48
3.5 Simulation study . . . 49
3.5.1 Simulated data generation . . . 49
3.5.2 Type I error rate for nonstationarity test . . . 50
3.5.3 Parameter estimation accuracy . . . 50
3.5.4 Prediction distribution accuracy . . . 51
3.6 Data application: Precipitation data in Puerto Rico . . . 51
3.6.1 May 2013 data . . . 52
3.6.2 January 2013 data . . . 53
3.7 Conclusion . . . 54
BIBLIOGRAPHY . . . 71
APPENDICES . . . 78
Appendix A Matrix Normal Distribution . . . 79
Appendix B Derivation of Posterior Distributions . . . 81
B.1 Posterior ofB∗ . . . 81
B.2 Posterior ofG. . . 82
B.3 Posterior ofΣ . . . 82
B.4 Posterior ofτ . . . 83
B.5 Posterior ofρ for CAR Model . . . 83
B.6 Posterior ofqii for DWT Model . . . 84
Appendix C Cross-validation Results . . . 85
Appendix D Covariance parameter MSEs . . . 88
LIST OF TABLES
Table 1.1 Cross-validation results, averaged over all prediction angles (10 - 80) . . . . 16
Table 1.2 Cross-validation results, averaged over interpolation angles (20 - 70) . . . . 16
Table 2.1 Accuracy of crystallographic parameters (I0,λ, 2θ0) . . . 38
Table 2.2 Accuracy of crystallographic parameters (U,V,W) . . . 39
Table 2.3 Coverage of crystallographic parameters (I0,λ, 2θ0) . . . 40
Table 2.4 Coverage of crystallographic parameters (U,V,W) . . . 41
Table 3.1 Estimation Accuracy for Simulated Data . . . 58
Table 3.2 Mean computation time (in seconds) for simulated data. . . 58
Table 3.3 Evaluation of Prediction Distributions for Simulated Data . . . 59
Table 3.4 Covariance parameter estimates for Puerto Rico precipitation data . . . . 61
Table 3.5 Five-fold cross-validation results for Puerto Rico precipitation data . . . . 62
LIST OF FIGURES
Figure 1.1 Example output for a molecular dynamics simulation . . . 3
Figure 1.2 Cross-validation results across all tilt tension angles. . . 17
Figure 1.3 Predictions from cross-validation for tilt angle 10 . . . 19
Figure 1.4 Predictions from cross-validation for tilt angle 50 . . . 20
Figure 1.5 Predictions from cross-validation for tilt angle 80 . . . 21
Figure 1.6 Predictive accuracy dimension reduction results for CAR and DWT models 22 Figure 2.1 True X-ray F1(2θ) and neutron F2(2θ) signals . . . 30
Figure 2.2 Absolute error between fitted and true X-ray signal for simulated data sets. 33 Figure 2.3 Absolute error between fitted and true neutron signal for simulated data sets. . . 34
Figure 2.4 Fitted X-ray background scattering for simulated data sets. . . 35
Figure 2.5 Fitted neutron background scattering for simulated data sets. . . 36
Figure 2.6 Fitted variable weighting functions for simulated data sets. . . 37
Figure 3.1 Means and standard deviations for Puerto Rico data . . . 55
Figure 3.2 Daily mean and max values for Puerto Rico data . . . 55
Figure 3.3 Maps of observed values for Puerto Rico data . . . 56
Figure 3.4 Longitude, latitude, and elevation across the island of Puerto Rico. . . 56
Figure 3.5 Simulated stationary and nonstationary data for two sites . . . 57
Figure 3.6 Type I error rate for nonstationarity test at 5% significance level . . . 60
Figure 3.7 Empirical correlograms for January 2013 and May 2013. . . 60
Figure 3.8 Fitted correlation functions for May 2013 . . . 63
Figure 3.9 Fitted correlation functions for January 2013 . . . 64 Figure 3.10 Predictions and standard deviations for 9 May 2013 usingp= 2 predictors. 65 Figure 3.11 Predictions and standard deviations for 9 May 2013 usingp= 4 predictors. 66 Figure 3.12 Predictions and standard deviations for 9 May 2013 usingp= 7 predictors. 67 Figure 3.13 Predictions and standard deviations for 26 Jan. 2013 usingp= 2 predictors. 68 Figure 3.14 Predictions and standard deviations for 26 Jan. 2013 usingp= 4 predictors. 69 Figure 3.15 Predictions and standard deviations for 26 Jan. 2013 usingp= 7 predictors. 70
Chapter 1
Spatial prediction of crystalline
defects observed in molecular
dynamic simulations of plastic
damage
Molecular dynamic computer simulation is an essential tool in materials science to study atomic properties of materials in extreme environments and guide development of new materials. We
propose a statistical analysis to emulate simulation output with the ultimate goal of efficiently
approximating the computationally-intensive simulation. We compare several spatial regression approaches including conditional autoregression (CAR), discrete wavelets transform (DWT),
and principal components analysis (PCA). The methods are applied to simulation of copper atoms with twin wall and dislocation loop defects, under varying tilt tension angles. We find
that CAR and DWT yield accurate results but fail to capture extreme defects, yet PCA better
captures defect structure.
1.1
Introduction
The permanent deformation of a crystalline material is typically accompanied by the creation
of defects that disturb the lattice structure. These defects can be in the form of partial planes of atoms (dislocations), atoms missing from lattice sites or in positions between sites (vacancies
and interstitials, respectively), regions of missing atoms (voids), boundaries across which the
lattice orientation changes (twins and grain boundaries), and regions in which the atoms are disordered (amorphous regions) [FA77].
Traditional modeling of plastic damage in metals involves numerically solving continuum
constitutive equations that relate structure, stress, strain and mechanical response. In recent decades, however, atom-scale approaches have started to play a larger role in modeling plastic
damage [Lu15]. In molecular dynamics simulations, for example, trajectories of individual atoms
are followed in time from an initial configuration by numerically integrating classical equations of motion that are coupled through a prescribed set of inter-atomic forces. Examination of the
trajectories, typically either through animations of the atom motion or by more quantitative
analyses, yields information about structures and their formation mechanisms as simulation conditions evolve. To study plastic deformation in a crystalline metal, for example, the motion
of collections of atoms can be followed as a system is strained.
The computational costs of a molecular dynamics simulation increase with an increasing number of atoms and with the length of time over which the atom motion is simulated. Because
of this, with current computing capabilities, simulations of plastic damage in crystals are typi-cally restricted to sub-micron length scales and sub-microsecond time scales, respectively, which
are far below practical engineering scales. Two of the leading challenges to molecular dynamics
simulations of this type are therefore making the simulations more efficient, and bridging de-fect formation mechanisms determined at the atomic scale with continuum, engineering-scale
constitutive relations [Bre13; MT09]. The results reported in this paper are a first step
to-ward exploring a unique response to these challenges in which coarse-grained representations of atomic configurations that evolve during plastic strain are connected through a statistical
model that is trained from a subset of fully atomistic simulations. Building prediction models
from computer simulation output is the focus of surrogate modeling [Sac89; Ken02], computer emulation [Bay07; Rou08], and meta-modeling [Che11; Wan14], but rarely have these methods
been applied in this manner to molecular dynamics.
To make a statistical analysis feasible, defect configurations from simulations with several hundreds of thousands of atoms are summarized by three-dimensional arrays of voxels that
contain information regarding defect types and densities (Figure 1.1). Our objective is to build
a statistical model to predict voxel profiles that correspond to atomic defect densities that would arise from the corresponding molecular dynamics simulation. We treat each array of
voxels as the functional response to the simulation conditionals, which frames the analysis
as a function-on-scalars regression problem. Methods for this kind of functional data analysis (FDA) have been explored at length in the past several years. General FDA methods have
been explored at length in [SR05] and [HK12], with typical approaches being either functional
Figure 1.1 Example output for a molecular dynamics simulation
Left: values for individual atoms, thresholded at 1 to indicate a molecular deformation around the atom. Right: 16×16×16 array of voxels with mean values, thresholded at 0 to indicate a molecular deformation within the voxel. Thresholds were chosen to clearly depict only poten-tially defective atoms/voxels that would be obscured by the clearly non-defective parts of the solid.
linear models [Cra09; He10], functional principal component analysis (FPCA) [Di09; Gre11], or nonparametric FDA [FV06]. FDA becomes more intricate when handling dependent functional
responses [Kok12], but a general approach is a functional additive mixed model [Sch14], which
flexibly accounts for a variety of correlation structures between functional responses. In our case, the functional response exhibits spatial dependence, which has been explored in [Del10], [Sta10],
and [Ram11]. Modeling this spatially-dependent functional response requires computationally
efficient representation of the spatial patterns to isolate those patterns that vary with functional factors.
In this paper, we explore statistical models to emulate results for molecular simulations of
defects created in strained metals. Our objective is to present and compare several approaches to this problem, evaluate the feasibility of applying statistical methods for this problem, and
offer recommendations for future development. We present a general approach to accommodate
both spatial variation across voxels and variation within a voxel across simulated conditions. While molecular dynamics includes temporal variation, we will focus on predicting the final state
of the simulation from more generalized conditions. For computational simplicity and ease of interpretation, we assume separability between the spatial variation and the simulation variation
[Gal94; NR01]. We project the simulation output into a spatially uncorrelated space using a
variety of spatial dependence structures: conditional autoregression (CAR) [Bes74], discrete wavelet transforms (DWT) [Mal89], and principal component analysis (PCA) [Hot33]. Variation
across design conditions is explained using semiparametric regression methods including Fourier
and Bernstein basis expansions. We utilize the matrix normal distribution [Daw81] to achieve a computationally efficient Markov chain Monte Carlo algorithm to fit the model. We apply the
methods to an atomic simulation of copper, evaluate the performance of the statistical model,
and provide recommendations for future research in this direction.
1.2
Data simulation and processing
1.2.1 Simulation generation
In molecular dynamics simulations atom trajectories are followed by numerically integrating
classical equations of motion that are coupled through a prescribed set of inter-atomic forces.
The simulations reported here were carried out using the LAMMPS software [Pli95], with interatomic forces from a many-body embedded-atom method potential for copper [Mis01].
The systems contained about 500,000 atoms in a periodic box with sides of roughly 1.6·10−8
meters; the numerical integration used a time step of 10−15 seconds. Initial atomic positions
for the grain boundaries were generated from two face-centered cubic lattices that were rotated by angles±θ along the<100>direction. The two lattices were then cut so that with periodic boundaries they met at two symmetric tilt grain boundaries in the direction perpendicular to the
applied strain, and so that the system satisfied the periodic boundaries in all three directions. Atoms were then removed that were less than 0.2 nm apart along the grain boundaries (for
reference the copper-copper near-neighbor distance is 0.254 nm) . In addition to the set of
symmetric tilt grain boundaries, separate systems with dislocation loops were also generated by removing slices of copper atoms from an ideal lattice. After the initial atomic configurations
were generated, the atom positions for each system were relaxed to minimize the potential
energy. This was followed by a constant-volume anneal at 300K for 100 ps.
To generate plastic damage, the systems were strained along the direction normal to the
grain boundaries (with no tilt this is the <100> direction) by scaling atom positions and the box length by a constant engineering strain up to a total strain of 0.1 over 100,000 time steps. This extremely rapid strain rate is a result of the computational limitations on system size and
time scale that are inherent to molecular dynamics simulations. While comparison to typical strain rates, which are order of magnitude slower, may be inappropriate, this high strain rate
is acceptable for the present study because of our goal of a statistical description of molecular
simulation results. To eliminate noise in the data, the atomic measures, such as the centro-symmetry parameter described in Section 1.2.2, were averaged over each 100 time steps during
the simulations.
1.2.2 Quantifying materials defects
While other atomic measures (e.g. common neighbor analysis, virial stress, and potential and kinetic energy) are possible defect summaries, we focus on the centro-symmetry parameter for
each atom,
CSP = 6
X
i=1
|Ri+Ri+6|2, (1.1)
where theRiterms represent directional vectors to the twelve nearest neighboring atoms, paired
such thatRi andRi+6have opposite directions [Kel98]. Compared to the other measures, CSP most efficiently measures the strength of defects in the crystal lattice. For a face-centered cubic lattice (which is the stable, ideal structure for copper), the ideal CSP value would be zero for
non-defective atoms and higher values for different defect types. Local fluctuations in atomic
structure due to vibrations in a molecular dynamics simulation can change these values, but the changes are sufficiently small that different types of defects can be clearly discerned. To eliminate
noise in the data from the simulation, the CSP values used in the analysis were averaged over each 100 time steps during the simulations. For this study, a logarithmic transform of the CSP
was used to allow for the normality assumptions made in Section 1.3.
1.2.3 Coarse graining
Coarse graining reduces the size of the data from several hundred thousand particles to a more manageable size. The spatial domain is divided into a cubic array of cells, or “voxels,” and
summary values are obtained for each cell. Coarse graining has often been used in molecular dynamics studies to simplify computation, such as polymer simulation [MP02] and biomolecular
systems [IV05]. Voxel-wise analysis is also used in medical imaging, particularly functional MRI
data [Wor02; Woo09], and uses projection methods to identify independent structures in the images [Lee11; Kaw12]. The number of grid cells should be large enough to capture important
structural features but small enough to permit efficient computing. Based on our exploratory
analysis and computing limitations discussed in Section 1.3.2.2, a 16 x 16 x 16 cubic grid sufficiently captured important spatial features. As the summary response, we use the mean
log-CSP for all particles in the voxel. Therefore the results of each simulation are represented
by n= 163 observations in a 3-D array, as seen in Figure 1.1. Other data summaries, such as the mean CSP, were attempted during the analysis but did not significantly affect results.
1.3
Model description
Let Y(s|θ) be the response in spatial cell s of the solid when the simulator is evaluated at L design features θ= (θ1, . . . , θL) . In our example, the L= 2 input values are tilt tension angle
(continuous) and the presence of a dislocation loop (binary) as discussed in Section 1.3.3. The responses are modeled using separable functions
Y(s|θ) =
p
X
k=1
βk(s)φk(θ) +e(s|θ), (1.2)
whereβk(s) are spatially-dependent coefficients,φk(θ) are fixed functions of inputs, ande(s|θ)
is the error. Therefore, variation in the response across design factors θ is regressed onto p basis functionsφk(θ), and the regression varies across the locationssvia the spatial coefficients
βk(s).
In vector form, the responses at n spatial locations s1, . . . ,sn for a given input θj are
expressed asYj = [Y(s1|θj), . . . , Y(sn|θj)]T. The simulator is evaluated atminputsθ1, . . . ,θm
to create the response matrix Y= [Y1. . .Ym]. The statistical model in (1.2) is written as
Y
n×m=nB×ppA×m+nE×m, (1.3)
where the elements ofB,A, andE are βk(si), φk(θj), ande(si|θj), respectively.
1.3.1 Prior Distributions
We use the matrix normal distribution [Daw81] to account for the dependence between and
within the rows and columns of the mean parameters B and errors E. See Appendix A for a full description of this distribution. The major assumption here is that the spatial covariance is separable from the design factor covariance,
Cov [e(s1|θ1), e(s2|θ2)] = CS(s1,s2)·CT(θ1,θ2), (1.4) Cov [βk1(s1), βk2(s2)] = CS(s1,s2)·CB(k1, k2), (1.5)
where CS, CT, and CB are covariance functions for space, design conditions, and basis
co-efficients, respectively. This assumption implies that the covariance between columns is the
same within each row and vice versa. While it may mask the interaction effect between specific
rows and columns, separability of the covariance structure drastically reduces the number of parameters we need to estimate.
For purposes of Bayesian inference, we apply a spatial model to the coefficients βj(s). The
mean of B is modeled using spatial predictorsx1(s), . . . , xq(s), such as the relative position of
the voxel within the solid. We use these predictors to model the expected value
E(B|X) = X
n×qqG×p, (1.6)
where the (i, j) element of X is xj(si) and G is a matrix of unknown regression coefficients.
Hence, we assume the following multivariate spatial prior distribution forB
B∼MatrixNormal(XG,U,Σ). (1.7)
Here the n×n matrix U is the spatial covariance matrix, whose elements are defined by CS(si,sj). The p×p matrix Σ is the covariance matrix for the coefficients within the same
voxel, whose elements are defined byCB(k1, k2).
The random errors inE are assumed to have mean zero. Furthermore, they are assumed to
be spatially correlated within each simulation and to be independent between simulations. The distribution of error matrixE is assumed to be
E∼MatrixNormal(0,U, σ2Im). (1.8)
The coefficient matrixBand error matrixEare assumed to have the same row covariance matrix U. The main purpose of this assumption is to project both B and E into the an uncorrelated space based on their assumed structure, as described in Section 1.3.2. This assumption may be relaxed such that the row covariances are similar enough to be projected using the same
projection matrix but have different parameter values to estimate.
To complete the model specification, we must select appropriate basis functionsφk, form
ap-propriate spatial covariance functionCS, and specify prior distributions for the hyper-parameters.
We apply the following prior distributions
G ∼ MatrixNormal(0,Φ,Σ),
σ−2 ∼ Gamma(a, b), (1.9)
Σ−1 ∼ Wishart(ν,Ψ),
where the matricesΦand Ψand the constants ν,a, and b are user-defined hyper-parameters. Different forms of the spatial covariance are considered in Section 1.3.2, and basis functions are
discussed in Section 1.3.3.
1.3.2 Spatial Projections
Because of the large number of voxels, analyzing the spatial model directly is not
computation-ally feasible. To allow for efficient computing, we project the data into a spaticomputation-ally uncorrelated
space. Let Pbe an invertible matrix such that PUPT =Q−1, where Q is a diagonal matrix. We project (1.3) usingPto get
Y∗ =B∗A+E∗, (1.10)
whereY∗=P Y,B∗=P B, andE∗ =P E. The projection accounts for the spatial correlation between cells, soB∗ and E∗ are distributed as
B∗ = ∼ MatrixNormal(X∗G,Q−1,Σ), (1.11) E∗ = ∼ MatrixNormal(0,Q−1, σ2Im),
where X∗ =P X. After this transformation, the only non-diagonal covariance matrix in (1.3) is Σ, which is typically low-dimensional. The exact form of Q will depend on the assumptions we place on U and P, and we explain the advantages and disadvantages of the various spatial projections in the subsequent sections.
1.3.2.1 Conditional Autoregressive Model
Assuming a process varies smoothly over the spatial region, an attractive model for the
spa-tial dependence between voxels is the conditional autoregressive (CAR) model [Bes74; HR10;
Ban14]. Modeling functional areal data using a CAR model has also been explored in [Zha15a]. Under the CAR model, the covariance matrix is U= (D−ρM)−1, whereM is the adjacency matrix for the voxels, D is a diagonal matrix containing the number of adjacent voxels on the diagonal, andρ∈(0,1) is the unknown spatial dependence parameter. The elements of the ad-jacency matrixMij equal one if voxelsi is adjacent tosj and zero otherwise, and the elements
of the diagonal matrix are Di =
Pn j=1Mij.
The CAR model regresses the response in a given voxel against the values in the adjacent voxels. For an arbitrary vector Z= (Z1, . . . , Zn)T, the conditional distribution ofZi under the
CAR model is normal with mean and variance
E(Zi|Z−(i)) = ρ
X
j6=i
Mij
Di
Zj, (1.12)
Var(Zi|Z−(i)) = σZ2 Di
. (1.13)
The variance parameterσ2Zis set to one because of confounding withσ2. The spatial covariance is determined by a single unknown parameter, ρ∈(0,1). Large ρ implies stronger dependence between voxels than small ρ. We restrict ρ to (0,1) so that U is positive definite, and assume priorρ∼Beta(c, d).
To obtain the orthogonal projection matrixPcorresponding toU, we compute the spectral decomposition of D−1/2MD−1/2 = ΓΩΓT such that Γ represents the orthonormal matrix of
eigenvectors andΩrepresents the diagonal matrix of singular values. Then
(D−ρM)−1= (ΓTD1/2)−1(In−ρΩ)−1(D1/2Γ)−1. (1.14)
Defining the projection matrix to be P=ΓTD1/2, the resulting form forQ is
Q=In−ρΩ. (1.15)
The values forD and M(and consequently Γand Ω) will be constants that can be reused. In Section 1.2.1, it was noted that the data simulations had periodic boundaries on the
domain. As a particle moved out of the domain, it would return immediately through the
opposite face. The voxels on the outer faces are hence related, so the adjacency matrix should reflect the periodic adjacency. The periodic conditions effectively nullify edge effects, which
makes the spatial model stationary and much easier to handle computationally.
1.3.2.2 Wavelet Decomposition
Assuming smooth spatial structure may not be ideal to identify localized defects, so we propose as an alternative the three-dimensional wavelet expansion for functional mixed models [MC06].
Instead of assuming a set structure for the spatial covariance, an arbitrary spatial processZ(s) is written as a wavelet series
Z(s) = X
j,k∈Z
Zjk∗ ψjk(s), (1.16)
whereψjk(s) are orthogonal wavelet basis functions obtained by scaling and shifting a generating
wavelet function. For example, if we start with the one-dimensional Haar wavelet [Haa10]
ψ(s) =
(
1, 0≤s≤1
0, otherwise, (1.17)
then a set of wavelet basis functions are
ψjk(s) = 2j/2ψ(2js−k). (1.18)
Higher dimensional wavelets are obtained through tensor products of the one-dimensional wavelets.
Because of the orthogonality, the wavelet coefficients can be calculated by integrating the
spatial process with the wavelet function
Zjk∗ =
Z
Rn
Z(s)ψjk(s)ds. (1.19)
The Zjk∗ coefficients are uncorrelated values that completely define the spatial process. After projecting the spatial process to its wavelet decomposition, we can use our model to predict
the coefficients without needing to model spatial covariances, then reverse the decomposition to obtain the predicted spatial process.
When the process is sampled at equally spaced points in the domain (such as our voxel array), the discrete wavelet transform (DWT) can quickly obtain the wavelet coefficients from
the observed responses using Mallat’s algorithm [Mal89]. The DWT reduces to applying an
orthonormal matrix P of the wavelet function values to the response vectors Y to obtain the wavelet coefficients Y∗. The row covariance matrix of Y∗ is the inverse of a diagonal matrix, Q, but each of then elements ofQ must be estimated during the MCMC sampling. We apply a Gamma prior to each diagonal element qii ∼ Gamma(c, d) of Q and use the wavethresh
package to implement the DWT for the three-dimensional data [Nas13].
Unlike the smoothness assumption in the CAR model, the DWT model allows for abrupt
local changes, which is potentially useful for finding smaller defects in the solid. The compu-tational drawback is that Mallat’s algorithm restricts the number of divisions in the coarse
graining. The algorithm requires that all dimensions have the same number of divisions and
that the number of divisions must be a power of two. This restriction may make adjusting to higher resolutions difficult since increasing the divisions on a side to the next power of two
would increase the total number of voxels by a factor of eight.
1.3.2.3 Principal Components
We also consider principal components analysis (PCA) [Hot33] to spatially project the data. The core of this method centers on empirically estimating the spatial covariance matrix from
the raw data and reducing it into the eigenvectors that account for the majority of the spatial
variability. If we assume that ˆG is an estimate of the mean coefficient matrix, then we can estimate the spatial covariance
ˆ U= 1
m
m
X
i=1
(Yi−XGaˆ i)(Yi−XGaˆ i)T, (1.20)
and then decompose the estimate into its singular value decomposition, ˆU = ΓDΓT. At the most, only m singular values in D will be non-zero. We use only the first m0 < m eigen-value/eigenvector pairs in the decomposition of ˆU.
Let Dm0 be a diagonal matrix of the m0 ≤m non-zero singular values and Γm0 be them0
associated eigenvectors. Like the projection for the CAR model, the projection matrix is P= D−m10/2ΓTm0. When applied to Y, the projected response Y∗ is an m0×m0 matrix, representing
a massive dimension reduction. Also, the rows of Y will be theoretically independent, as in Q = Im0, so only the covariance matrix of the columns needs to be modeled. We use all m
non-zero singular values for this application, sincem is small in this case.
The estimate for ˆU needs to be determined for each design set-up, so the projection matrix cannot be calculated independently of the data, as in the CAR or DWT models. The estimate
ˆ
B affects ˆU as well. We use the least-squares estimate of the spatial coefficients ˆ
G= (XTX)−1XTYAT(AAT)−1, (1.21) and calculate ˆUonce instead of for each MCMC sample. The ˆUmatrix can reasonably capture the complex features of the spatial covariances, provided thatm is moderately large.
1.3.3 Basis Functions of Input Space
For the current data set, two design factors under investigation are the tilt tension angle and
the presence of a dislocation loop in the initial configuration. The tilt tension angle, denoted
asα, is defined as the acute planar angle formed by the atomic planes of the lattices along the grain boundaries. A dislocation loop is formed by removing slices of copper atoms from an ideal
lattice, as described in Section 1.2.1.
The tilt angle is a continuous input variable on [0,90] since any tilt angle larger than 90 degrees would be equivalent to one smaller than 90 degrees. We need to define the functions of
angle to be used in the basis expansion in (1.2). We restrict the basis functions to be continuous
functions inL2on the [0,90] degree domain. The angle functions could be periodic on the [0,90] degree domain because any planar angle larger than 90 is equivalent. As a result, we consider
the Fourier basis functions, which use trigonometric functions of different frequencies
ϕj(α) =
cos j π 90α
, j = 1, . . . , J sin
(j−J)π 90 α
, j=J + 1, . . .2J
(1.22)
where the number of angle functions is K = 2J. While the Fourier basis models periodic behaviors over the entire angle domain, it may have trouble modeling more localized deviations
from the overall behavior. We consider the Bernstein polynomials as an alternative basis. For
a fixed degreeJ, the Bernstein basis polynomials on the domain [0,90] are
ϕj+1(α) =
J j α 90 j
1− α
90
J−j
, j = 0, . . . , J. (1.23)
where the number of angle functions isK =J + 1. We include a constant function, ϕ0(α) = 1 to allow for effects that are constant over tilt angle.
The dislocation loop is a small plane of atoms removed from one of the lattices. Some of the simulations had the dislocation loop while others did not. We define two factors
ψ1(θ) = 1, ψ2(θ) =
(
0, if no dislocation loop exists in initial solid
1, if dislocation loop exists in initial solid (1.24)
that combine with the tilt angle functions to express the design factors in (1.2). Thus, the design factors are
φk(θ) =
(
ψ1(θ)ϕk−1(α), k= 1, . . . , K + 1
ψ2(θ)ϕk−K−2(α), k=K+ 2, . . . ,2K+ 2
(1.25)
where the number of functions is p= 2K+ 2.
1.4
Computing Details
We use Markov chain Monte Carlo sampling to evaluate the posterior distribution. The Gibbs
sampling algorithm begins with initial values for all model parameters and then sequentially
draws each parameter from its full conditional distribution to yield posterior samples. We give the full conditional distributions below.
Under the spatial projections for B and E and the prior distributions (1.9), we derive the full conditional distributions of the parameters (see Appendix B for derivations) to be
(B∗|Y∗,G,Σ, τ,Q)∼MatrixNormal
MB∗,Q−1,VB∗, (1.26)
(G|Y∗,B∗,Σ, τ,G)∼MatrixNormal [MG,UG,Σ], (1.27) (Σ|Y∗,B∗,G, τ,G)∼InverseWishart
ν+n+q,Ψ+GTΦ−1G+SSE(B∗)
,(1.28) (τ|Y∗,B∗,G,Σ,Q)∼Gamma
a+mn 2 , b+
1
2 ·tr[SSE(Y ∗
)]
, (1.29)
whereτ =σ−2 and
MB∗ = τY∗AT +X∗GΣ−1VB∗, VB∗ = (τAAT +Σ−1)−1, (1.30)
MG = UGX∗TQB∗, UG=
X∗TQX∗+Φ−1
−1
, (1.31)
SSE(Y∗) = (Y∗−B∗A)TQ(Y∗−B∗A), (1.32) SSE(B∗) = (B∗−X∗G)TQ(B∗−X∗G). (1.33)
Samples for these parameters can be obtained from (1.26)-(1.29) using Gibbs sampling. How-ever, posterior samples forQ depend on how the projections are performed.
Under the conditional autoregressive model, Q depends on the propriety density ρ, which has full conditional
π(ρ|Y∗,B∗,G,Σ, τ)∝ ρ
c−1(1−ρ)d−1
|In−ρΩ|−(m+p)/2
exp
(
−tr[τ SSE(Y
∗)] +tr
Σ−1SSE(B∗) 2
)
. (1.34)
Unfortunately, the density does not conform to a known distribution. Sampling from the pos-terior distribution of ρmust be done using Metropolis-Hastings. If we select a candidate value ρ0 from the current valueρtusing the distribution π(ρ0|ρt), we calculate the acceptance ratio
A(ρt, ρ0) =
π(ρ0|Y∗,B∗,G,Σ, σ2) π(ρt|Y∗,B∗,G,Σ, σ2)
·π(ρt|ρ
0)
π(ρ0|ρ
t)
, (1.35)
and from an independently generated U ∼Uniform(0,1), the next sampled value is ρt+1=ρt·IU > A(ρt, ρ0)
+ρ0·I
U ≤A(ρt, ρ0)
. (1.36)
We reduce autocorrelation between MCMC samples ofρby Stochastic thinning via a Geometric resolvent [RC13]. This method involves generatingL from a Geometric distribution and using (1.36) to obtainρt+L, which is saved as the MCMC sample for that iteration.
Under the wavelet decomposition, the posterior distribution of each element Qis
(qii|Y∗,B∗,G,Σ, τ,Q−(ii))∼Gamma
c+ m+p 2 , d+
τ 2ke
(i)
Y∗k2+
1 2ke
(i)
B∗k2
Σ−1
, (1.37)
whereke(Yi)∗kis theL2-norm of theithvector of (Y∗−B∗A) andke(Bi)∗kΣ−1 is theΣ−1-weighted
L2-norm of theithrow vector of (B∗−X∗G). See Appendix B.6 for the full derivation. We use Gibb’s sampling to estimate each element ofQ, which is a major advantage of using DWT over the CAR model.
1.4.1 Reducing dimension to improve computation time
Typically the number of simulations will be much smaller than the number of voxels. As a result, the PCA-projected matrices will be much smaller than the projected matrices for the
CAR or DWT models. The smaller matrices would reduce computation times for the repeated
matrix multiplications, which is the major advantage of using PCA over the other two methods.
To enhance the computation speed of the other projection methods, we reduce the size of the projected matrices by removing rows that are near zero across all design inputs. The sum
of the squared values are computed across the rows of Y∗ and then ranked. The rows with the largest sum of squares are included in Y∗ as usual while the rest of the rows are set at zero, so only the corresponding rows ofB∗ need to be updated. We will explore how dimension reduction affects predictive accuracy in Section 1.5.
1.4.2 Prediction Distribution
A major goal of this investigation is to predict the response under certain inputs. LetA0 be the matrix of design factors at which one wants to predict the responseY0. Since ˆY0 =BA0+E0, whereB∼MatrixNormal(XG,U,Σ) and E0 ∼MatrixNormal(0,U, σ2I), we can derive
ˆ
Y0 ∼MatrixNormal(BA0,U,AT0ΣA0+σ2I) (1.38) to be the predictive distribution. The predictive distribution is used to evaluate the model using
cross-validation, leaving out a design input to obtain estimates for the parameters, predicting the response at the removed input, and comparing the predictions to the observed simulation
results. Prediction intervals for each predicted value are developed using (1.38)
( ˆY0)ij ±zc/2
q
Uii·(AT0ΣA0+σ2I)jj, (1.39)
wherezc/2 is the critical value for the Standard Normal distribution.
1.5
Results
We use cross-validation to compare methods. For each fold, we withhold all simulations for one tilt angle, which is a total of three simulations including the two with dislocation loops and
the one without it. The remaining simulations are used to estimate the model and compute
predictions on the withheld simulations. We measure predictive accuracy using the predictive mean squared error
\ M SE= 1
n|R| n
X
i=1
X
r∈R
(Yir−Yˆir)2, (1.40)
wheren is the number of voxels,R is the set of removed simulations used for cross-validation, Yij is the observed log-CSP, and ˆYij is the predicted log-CSP. We also calculate the correlation
between the observed and predicted values to measure how well the models reproduce the defect
structure as well as the coverage of the 95% prediction intervals. For each model, we select weak prior distributions for the hyper-parameters1 to make the results data driven.
Table 1.1Cross-validation results, averaged over all prediction angles (10 - 80)
Projection CAR DWT PCA
Expansion Fourier Bernstein Fourier Bernstein Fourier Bernstein MSE 0.2690 0.2772 0.2876 0.3389 0.4792 0.3877 Coverage 0.8057 0.7944 0.9144 0.9081 0.9977 0.9926 Correlation 0.4084 0.4080 0.3923 0.3914 0.3364 0.3395
Table 1.2Cross-validation results, averaged over interpolation angles (20 - 70)
Projection CAR DWT PCA
Expansion Fourier Bernstein Fourier Bernstein Fourier Bernstein MSE 0.2156 0.2155 0.2230 0.2294 0.2999 0.2812 Coverage 0.7991 0.7896 0.9099 0.9065 0.9998 0.9975 Correlation 0.4669 0.4807 0.4444 0.4598 0.4002 0.4077
First, we consider the cross-validation results for the models without using the dimension
reduction technique described in Section 1.4.1. Results are plotted in Figure 1.2 and appear in tables in Appendix C. Table 1.1 contains averages over all tilt angles, and Table 1.2 contains
averages over angles 20 through 70. In terms, the CAR projection with the Fourier expansion
predicts the log-CSP values most accurately. The coverages, however, indicate that the CAR model may not have adequate inferential properties, as only about 80% of the prediction
in-tervals contain the true value. The DWT projections are fairly accurate with moderately good
inferential properties, as indicated by the low MSE and coverages closer to the desired con-fidence. PCA projections yield inaccurate predictions that have low correlation with the true
values, but the prediction intervals have near perfect confidence.
Across all models, the predictive accuracy decreases when predicting at tilt angles 10 and
1Φ= 100·I
q,Ψ= 10·Iq,ν=p+ 2, anda=b= 0.1. For the DWT model, we setc=d= 0.1. For the CAR model, we setc=d= 0.1 and define adjacency as voxels that share a face, edge, or diagonal corner.
(a)Mean Squared Error
(b) Coverage (c) Correlation
Figure 1.2 Cross-validation results across all tilt tension angles.
80. This behavior is most likely be a result of these angles being on the outer edges of the domain. Prediction at these values would be extrapolation of the training data set, which is
generally less reliable than interpolation at the inner tilt angles.
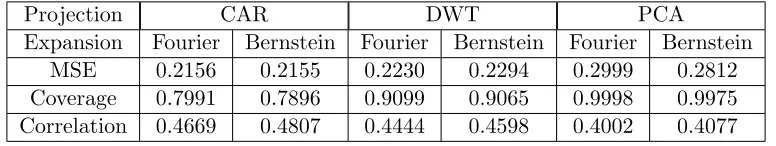
Next we visually inspect three-dimensional slice plots of cross-validation predictions. Figure 1.3 contains the observed and predicted values for tilt angle 10. The CAR and DWT models
do not capture the true values, predicting heavy defects throughout the lower half of the solid
but underestimating defects in the upper half. The twin-wall defect is not even apparent in the DWT projection with Bernstein polynomials. The PCA model overestimates defect intensity
but accurately locates defect structures.
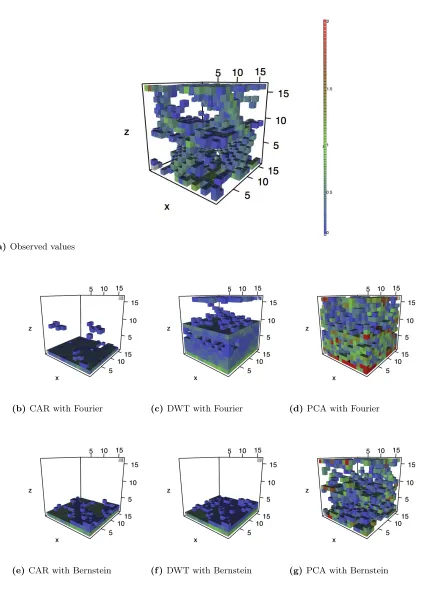
Figure 1.4 contains the observed and predicted values for tilt angle 50. The CAR model depicts some dislocations in the defect structures but smooths the intensity over the neighboring
voxels, particularly in the Bernstein expansion. DWT model appears biased towards predicting
the non-defective voxels that make up a large part of the solid, as only the twin-wall defect can be seen. PCA once again overestimates defect intensity, but dislocations and twin wall defects
are clearly represented.
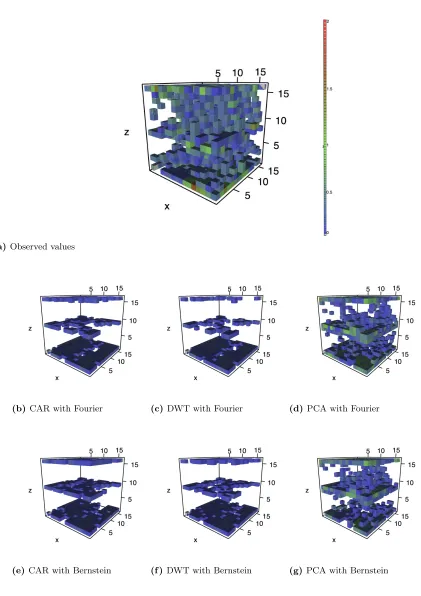
Figure 1.5 contains the observed and predicted values for tilt angle 80. Similar to the
behav-ior in tilt angle 10, the CAR and DWT models fail to accurately predict the dislocation defects,
though here they predict heavy defects in the upper half of the solid instead of the lower half. Similarly, the PCA model overestimates defect intensity but still locates the defect structures
throughout the solid.
Over all the tilt angles, the three-dimensional slice plots indicate that the PCA model is consistently better at predicting the location of the defect structures. The CAR and DWT
models assume that the data is spatially smoother than it actually is, leading to predictions
that fail to depict the dislocation defects.
Finally, we consider how dimension reduction in the CAR and DWT models affects the
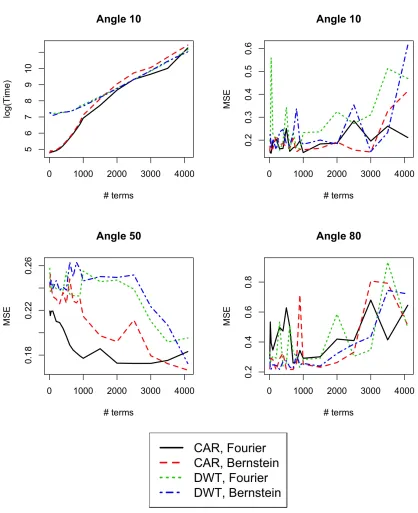
computation and prediction accuracy. Figure 1.6 plots computation time and the predictive
MSE against the number of terms retained in the model. Computation increases log-linearly in the number of terms, and the DWT models require generally more computation than the CAR
models. From the predictive MSE values, we see that the predictive accuracy either does not
improve or even decrease by having more model terms, particularly for the DWT models. More terms in the model leads to overfitting of the training data, which can exacerbate extrapolation
errors. Conversely, the predictive accuracy at the interpolation angles improves with more terms,
but the CAR model does not need as many terms as the DWT model to significantly improve the MSE.
(a)Observed values
(b) CAR with Fourier (c) DWT with Fourier (d) PCA with Fourier
(e)CAR with Bernstein (f )DWT with Bernstein (g) PCA with Bernstein
Figure 1.3 Predictions from cross-validation for tilt angle 10
Colored voxels distinguish defects due to high log-CSP values. Voxels with values below zero
(a)Observed values
(b) CAR with Fourier (c) DWT with Fourier (d) PCA with Fourier
(e)CAR with Bernstein (f )DWT with Bernstein (g) PCA with Bernstein
Figure 1.4 Predictions from cross-validation for tilt angle 50
Colored voxels distinguish defects due to high log-CSP values. Voxels with values below zero
(a)Observed values
(b) CAR with Fourier (c) DWT with Fourier (d) PCA with Fourier
(e)CAR with Bernstein (f )DWT with Bernstein (g) PCA with Bernstein
Figure 1.5 Predictions from cross-validation for tilt angle 80
Colored voxels distinguish defects due to high log-CSP values. Voxels with values below zero
Figure 1.6 Predictive accuracy dimension reduction results for CAR and DWT models
Horizontal axis indicates the number of rows of the projected data used in the training dataset, ranging from 10 to 163. Top-left figure shows the natural log of the computation time (in seconds) for angle 10 changing linearly with the number of terms; similar behavior is observed for other tilt angles.
1.6
Conclusion
We explored spatial functional models to predict the output images of molecular simulations.
Our objectives were to assess whether this is feasible and to evaluate the strengths and weakness of various approaches. For simulations involving interpolating predictions (angles 20 through
70), the mean squared error between predicted and observed was approximately 0.25, with
correlation 0.45. For simulations involving extrapolating predictions (angles 10 and 80) the mean squared error was significantly higher with lower correlation. Therefore, there is promise
that statistical prediction can be a useful supplement to numerical simulation for interpolation
purposes.
Of the spatial projection methods, PCA would be the preferred method for predicting plastic
damage. Despite being less accurate in terms of mean squared error, PCA captures the localized defects, which are by nature extreme values in the solid. CAR is consistently accurate even while
extrapolating, but the prediction intervals do not obtain their desired confidence. DWT leads to
fairly accurate prediction results with adequate inferential properties, but underestimates the defect structure in the lattice. For both CAR and DWT, dimension reduction vastly reduces
the computation time and may reduce errors in the prediction due to overfitting.
Further work could also explore the variation structures in the design conditions. This analysis focused solely on the final images of independent simulations using a single design
factor, but the model allows for more complex dependencies between the images. It would be
of particular interest to explore how the images change over the course of a single simulation, using the initial image to predict the probable final image.
In addition, analyzing the quantiles of the CSP distribution may be an interesting approach
to modeling material defects. Defective molecules have a high CSP but represent a small pro-portion of the total solid, so estimates and accuracy measures based on the mean may be
misleading. Therefore, a model for the upper quantiles of the CSP values in each voxel may
outperform a model for the average log-CSP. This extreme value analysis requires specialized techniques and distributions (such as the generalized Pareto distribution) but may improve
upon the predicted defect structures offered by the PCA spatial projections.
Chapter 2
Bayesian estimation of
crystallographic parameters through
weighted fusion of radiation
diffraction data
We develop a Bayesian model to accurately estimate crystallographic parameters from radiation diffraction data, an important concern in the field of crystallography and materials science.
Our model integrates measurements from multiple diffraction data sources using weighted data
fusion. Each diffraction data source is decomposed into the Bragg’s signal and background noise. The error has heterogeneous variance conditioned on the strength of the signal and a
variable weighting function that is estimated through Markov chain Monte Carlo (MCMC). This weighting function reduces the influence of less reliable or corrupted diffraction data,
leading to more accurate estimates of the crystallographic parameters. We perform a simulation
study that shows how variable weighting in the data fusion of X-ray and neutron diffraction significantly improves the crystallographic parameter estimates, particularly when we simulate
corruption in the data.
2.1
Introduction
In crystallography, diffraction is a powerful tool for identifying the atomic structure of materials.
Radiation waves, composed of either X-rays or neutron waves, are directed on a crystal lattice
and diffracted by the atoms. Bragg’s Law [BB13] predicts the diffraction angles given the value of various parameters about the lattice structure. Over the last century, efforts have been made
to observe and catalog the diffraction pattern for a variety of crystal and powder structures.
These efforts have led to massive databases of cataloged materials, such as the Materials Genome Initiative [Pab14].
The Rietveld Method [Rie69] minimizes the relative squared differences the observed and
theoretical scattering parameters. This method is the standard approach to estimating crystal-lographic parameters and has been implemented through software like the General Structure
Analysis System [LVD86; TVD13]. However, this method has been shown to be subject to errors
related to false minima and uncertainty quantification [Wil06]. More recently, work has been produced on applying Bayesian statistical modeling of atomic parameters from X-ray diffraction
data [BM11; GL16; Fan16].
Materials scientists are now commonly faced with the statistical challenge of coherently combining data from multiple sources, such as data of different measurement types or data
collected under different environmental conditions and different measurement devices. As a specific and important special case, we incorporate both X-ray and neutron diffraction data into
a Bayesian model to improve the estimation of the crystallographic parameters. Each radiation
source has its advantages and disadvantages [Est14], but combining the sources improves the estimates overall [Ush12; GL15; Zha15b].
For these data, we observe intensities over a range of scattering angles for two sets of
diffraction data,Y1(2θ) andY2(2θ), from two experiments on a crystal or powder material. The material of interest can be described bydcrystallographic parametersα= (α1, . . . , αd)T. These
diffraction signals are measured at n1 and n2 angles, respectively. The standard approach to estimating crystallographic parameters using data fusion minimizes
n1
X
i=1
W1(2θi)
[Y1(2θi)−Yˆ1(2θi;α)]2
Y1(2θi)
+
n2
X
i=1
W2(2θi)
[Y2(2θi)−Yˆ2(2θi;α)]2
Y2(2θi)
, (2.1)
where ˆY is the expected intensity for a material with crystallographic parametersα. In (2.1), Yk appears in the denominator to adjust for the larger variance for angles with higher intensity.
The weights Wk(2θi) determine the degree to which the measured intensity Yk at angle 2θi
affects the estimate forα. Naturally, ifWk(2θi) = 0, then the diffraction measure at that angle
is not used to estimate the crystallographic parameters. The weights are often taken to be constant for all angles,Wk(2θ) =wk. The user determines the weightsw1 and w2 based on the source that is deemed most reliable, and sensitivity to this choice is informally explored.
Here we present a method of estimating the weights for each source, yielding a data-driven fusion of the diffraction measures. The resulting estimated weight functions allow reliability
comparisons of the diffraction data intensities, both within each source and between the different
sources. In the following sections, we develop a Bayesian model that obtains both probabilistic estimates for the crystallographic parameters and weighting functions based on the uncertainty
of the data. This method is then used on simulation data to show how it improves the accuracy
of parameter estimates.
2.2
Statistical Model
We decompose thekth response into the signal Fk, the background scattering Bk, and random
errorEk
Yk(2θ) =Fk(2θ;α) +Bk(2θ) +Ek(2θ|α). (2.2)
The signal Fk depicts the true diffraction intensities as given by Bragg’s Law for given
crystal-lographic parametersα. The crystallographic parameters αare shared by all data sources, but
the data sources have separate background functions and independent errors.
The background scattering Bk(2θ) captures other signals in the mean that are not directly
related to the crystallographic parameters. The background is approximated as the linear
com-bination ofp spline functionsXj(·)
Bk(2θ) = p
X
j=1
Xj(2θ)βkj, (2.3)
with the prior assumption βkj iid
∼ Normal(0,1/φk) for the coefficients. Here we have assumed
the same basis expansion for all data sources, but this model can be easily generalized to have
a unique basis for each source.
After accounting for the signal and background scattering, the error process Ek(2θ) is
as-sumed to be independent overkand 2θ. As the intensities are count data, a Poisson distribution is a natural choice for the model [e.g. [BM11]], but the counts in crystallography are large enough to permit a normal approximation. Therefore, we assume the errors are Gaussian with mean
zero and variance proportional to the signalFk. The variance is
Var[Ek(2θ)|Fk(2θ;α)] =σ2k(2θi) =τk−1
h
1 +eδkF
k(2θ;α)
i
eGk(2θ), (2.4)
where τk−1 is the overall variance and eδk scales the signal’s effect on the variance. A one is
added toeδkF
k to avoid having zero variance at angles with no signal.
The function Gk(2θ) allows for heterogeneous variance across the diffraction angles. For
example, we might expect Gk(2θ) to be larger when there exists a discrepancy between the
signal and the data, and this model misspecification inflates the variance. To estimate the heterogeneous variance,Gk is expressed as the linear combination of q basis functions
Gk(2θ) = q
X
j=1 ˜
Xj(2θ)γkj (2.5)
as in (2.3). To ensure identification with the overall variance, we select basis functions with ˜
Xj(0) = 0 for alljand thuseGk(0) = 1, uniquely identifyingτk−1 as the variance for angles near
zero.
Under this model, twice the negative log-likelihood for a single source is
nk
X
i=1
logσk(2θi) +σk−2(2θi)[Yk(2θi)−Fk(2θi;α)−Bk(2θi)]2 .
For comparison with the data fusion equation in (2.1), this can be rewritten as
nk
X
i=1
logσk(2θi) +Wk(2θi)
[Yk(2θi)−Fk(2θi;α)−Bk(2θi)]2
[1 +δkFk(2θi;α)]
.
where
Wk(2θ) =e−Gk(2θ) (2.6)
measures the precision of data source k at angle 2θ. As a result, twice the joint negative log-likelihood of the X-ray and neutron data
n1
X
i=1
logσ1(2θi) +W1(2θi)
[Y1(2θi)−F1(2θi;α)−B1(2θi)]2
[1 +δ1F1(2θi;α)]
+ n2 X i=1
logσ2(2θi) +W2(2θi)
[Y2(2θi)−F2(2θi;α)−B2(2θi)]2
[1 +δ2F2(2θi;α)]
(2.7)
which resembles (2.1) with Wk(2θ) as the weight function. This weighting function effectively
decreases the influence of angles with high error variance, whether it results from natural
variability in the intensity counts or through model misspecification.
2.3
Computational Details for Parameter Estimation
In order to perform Markov chain Monte Carlo (MCMC) and obtain estimates of the
crystallo-graphic parameters, we must first derive the posterior distributions for the model parameters. First, we express the model in Equation (2.2) in the following vector form
Yk=Fk+Bk+Ek (2.8)
where
Yk = [Yk(2θ1), . . . , Yk(2θnk)]
T;F
k= [Fk(2θ1;α), . . . , Fk(2θnk;α)]
T,
Bk = [Bk(2θ1), . . . , Bk(2θnk)]
T;E
k = [Ek(2θ1), . . . , Ek(2θnk)]
T.
Equation (2.3) is expressed asBk=Xβk, wherexi = [X1(2θi), . . . , Xp(2θi)]T,X= (xT1, . . . ,xTn)T,
and βk= (βk1, . . . , βkp)T.
2.3.1 Background and Precision Parameters The full conditional distribution forβk is
βk|rest∼Normal
(
V
n
X
i=1
Wk(2θi)[Yk(2θi)−Fk(2θi;α)]xi,V
) (2.9) where V= " n X i=1
Wk(2θi)xixTi +φkIp
#−1
. (2.10)
For the precision parameters, we assume the prior distribution τk, φk∼Gamma(a, b), yielding
the following full conditional distributions:
τk|rest ∼ Gamma
"
a+nk 2 , b+
1 2
X
i
[Yk(2θi)−Fk(2θi)−xTi βk]2e−Gk(2θi)
1 +eδkFk(2θi)
#
(2.11)
φk|rest ∼ Gamma
"
a+p 2, b+
P
jβkj2
2
#
. (2.12)
We obtain posterior samples ofδk andγkj using a random-walk Metropolis-Hastings algorithm
with Gaussian candidate distributions and regular tuning for an the acceptance rate between 20% and 50%.
2.3.2 Crystallographic Parameters
For the crystallographic parameters, we denote the range of physically plausible values for αl
as [Ll, Ul]. In the examples considered here, we have two cases: bounded parameters with finite |Ll| and |Ul|, and one-sided bounds with |Ll| < ∞ and Ul = ∞. In the first case, we select
a Uniform prior, αl ∼ Uniform(Ll, Ul), and in the second case, we select a log-Normal prior,
log(αl−Ll)∼Normal(0,1). For MCMC sampling, we transform the crystallographic parameters
αl into normally distributed latent variablesZl using the transformations
Zl=
Φ−1
αl−Ll
Ul−Ll
, Ul <∞
log(αl−Ll), Ul =∞
. (2.13)
where Φ−1is the inverse cumulative density function of the normal distribution. After obtaining
posterior estimates for the latent variables, we transform Zl back into the original
crystallo-graphic parameter
αl=
(
Ll+ (Ul−Ll)Φ(Zl), Ul<∞
Ll+ exp(Zl), Ul=∞
(2.14)
where Φ is the cumulative density function of the normal distribution. We use a random-walk Metropolis-Hastings algorithm to obtain posterior samples ofZl with Gaussian candidate
distributions and regular tuning for an the acceptance rate between 20% and 50%.
2.4
Simulation Study
The simulation study explores two questions of interest regarding crystallography. First, we
explore whether the fusion of both X-ray and neutron diffraction data significantly improves estimation of crystallographic parameters compared to using only one data source. Second, we
assess the ability of the proposed data fusion approach to identify reliable information and its
performance gain over more naive fusion approaches.
2.4.1 Data Generation
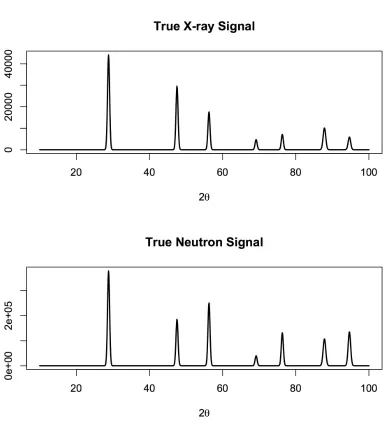
To generate the data, we set the crystallographic parameters to fixed values and generate a
synthetic signal for both the X-ray and neutron data. The synthetic signal Fk is calculated
from
Fk(2θ) =I0
X
hkl
SFhklL(2θ)H(λ,2θ0, U, V, W) (2.15)
Figure 2.1 True X-rayF1(2θ) and neutronF2(2θ) signals
Signals used in the simulation study, plotted by scattering angle 2θ.
where L(2θ) represents the Lorentz-polarization correction and SFhkl represents the structure
factors of gold [Fan16]. The vector α contains six parameters: incident intensity (I0),
wave-length (λ), angle offset (2θ0), and Caglioti parameters (U,V, andW). For this study, data was generated using I0 = 101, λ= 1.53, 2θ0 = 0.57, U = 7·10−7, U =−3·10−5, and W = 0.04. The resulting curves are plotted in Figure 2.1.
For the simulated X-ray data, we use the following functions for the true background function
B1 and heterogeneous variance function G1
B1(2θ) = 5 4cos 2θ 4 +2θ 25sin 2θ 3 , G1(2θ) =
2θ 30cos 2θ 4 +2θ 40sin 2θ 3 .
For the simulated X-ray data, we use the following functions for the true background function B2 and heterogeneous variance function G2
B2(2θ) = 100 2θ cos 2θ 4
+ 2 sin
2θ 3
, G2(2θ) =
10 2θcos 2θ 4 +60 2θsin 2θ 3 .
We construct the error variance using Equation (2.4), withτ = 5 andδ = log(0.001).
To study the model’s ability to identify reliable information, we simulate corruption in the data using an incorrect mean with signal set to zero for some angles. For X-ray signals, angles
in the upper third of the domain were corrupted
F1C(2θ;α) =
(
F1(2θ;α), 2θ <66.7
0, otherwise. (2.16)
For neutron signals, angles in the lower third of the domain were corrupted
F1C(2θ;α) =
(
F1(2θ;α), 2θ >33.3
0, otherwise. (2.17)
We simulate data with corruption in both, one, or neither data source. For each of these
sce-narios, we generate 100 data sets. Each simulated data set is fit using either X-ray data only,
neutron data only, or both data sources. We also fit the data either allowing the weighting function to vary across angle or be constant (i.e. Gk(2θ) = 0 for all θ). For all models, we
use p= 20 spline basis functions for the background. For models with varying weight, we use q = 10 spline basis functions to fit Gk, with the restriction that Gk(0) = 0. For each fit of the
simulated data, we generate 2000 MCMC samples and discard the first 1500 as burn-in.
2.4.2 Results and Discussion
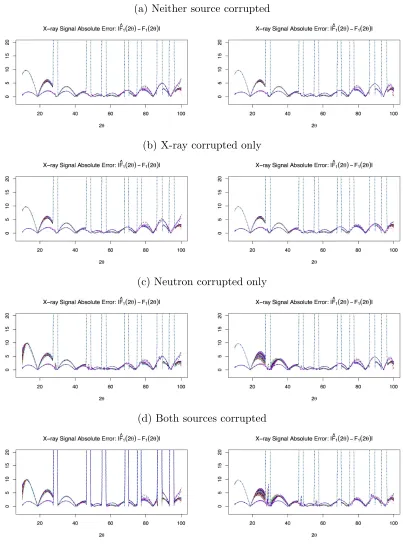
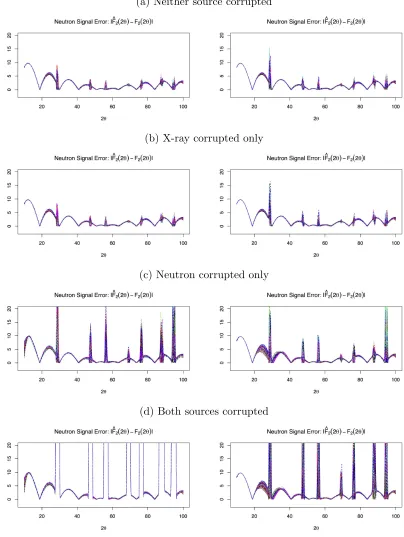
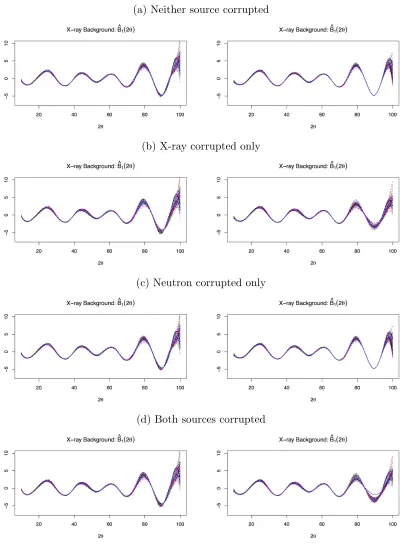
Figures 2.2 and 2.3 contain plots of the posterior means for the signal absolute error|Fˆk(2θ)−
Fk(2θ)|. Figures 2.4 and 2.5 contain plots of the posterior means for the background scattering
ˆ
Bk(2θ). Figure 2.6 contains plots of the posterior means for the log of the weighting functions
logWk(2θ) =−Gˆk(2θ). For these plots, we considered either both sources corrupted or neither
source corrupted. Each data set was fit using both sources, with either constant or varying
weights. These figures illustrate how the weighting function reacts to data corruption and affects the estimated background scattering and signal. When the data are corrupted, the signal’s effect
on the parameter estimate is decreased overall, as shown by the scales in the weighting functions.
TheGkfunction compensates for the decreased signal effect, but the varying weight has greater
freedom to adjust. The varying weight decreases around the corrupted angles, but the constant
weight decreases over all of them. The estimated background scattering has higher variability at
angles with lower weight, so the varying weight function leads to better fits of the background for both corrupted and uncorrupted data sources. As a result, the varying weight leads to less
absolute error in the signal, particularly at the peaks.
Tables 2.1 and 2.2 contain means and standard errors for squared relative errors for the crystallographic parameters estimates. The squared relative error is calculated as
( ˆαl−αl)2
α2
l
(2.18)
where ˆαl is the posterior mean and αl is the true value used in the data generation. Tables
2.3 and 2.4 contains coverage proportions of 95% credible intervals for the crystallographic
parameters.
Overall, the data fusion model with varying weights offers consistently accurate parameter
estimates. The MSE and coverages for the fusion model with varying weights are competitive
with the best models across all scenarios. When the neutron data are not corrupted, the model with only neutron data and constant weight has the smallest MSE values, but if it is corrupted,
the MSE and coverages for the parameters for this model are significantly worse than the other models, particularly for the initial intensity I0 and Cagliotti parametersU,V, and W. For the X-ray models, the varying weights always lead to improved accuracy in the MSE.
(a) Neither source corrupted
(b) X-ray corrupted only
(c) Neutron corrupted only
(d) Both sources corrupted
Figure 2.2 Absolute error between fitted and true X-ray signal for simulated data sets.
Lines represent fitted function from each simulation based on the posterior means. Left side plots used constant weighting; right side plots used variable weighting.
(a) Neither source corrupted
(b) X-ray corrupted only
(c) Neutron corrupted only
(d) Both sources corrupted
Figure 2.3 Absolute error between fitted and true neutron signal for simulated data sets.
Lines represent fitted function from each simulation based on the posterior means. Left side plots used constant weighting; right side plots used variable weighting.
(a) Neither source corrupted
(b) X-ray corrupted only
(c) Neutron corrupted only
(d) Both sources corrupted
Figure 2.4 Fitted X-ray background scattering for simulated data sets.
Lines represent fitted function from each simulation based on the posterior means. Left side plots used constant weighting; right side plots used variable weighting.
(a) Neither source corrupted
(b) X-ray corrupted only
(c) Neutron corrupted only
(d) Both sources corrupted
Figure 2.5 Fitted neutron background scattering for simulated data sets.
Lines represent fitted function from each simulation based on the posterior means. Left side plots used constant weighting; right side plots used variable weighting.
(a) Neither source corrupted
(b) X-ray corrupted only
(c) Neutron corrupted only
(d) Both sources corrupted
Figure 2.6 Fitted variable weighting functions for simulated data sets.
Lines represent fitted function from each simulation based on the posterior means. Left side plots are for X-ray diffraction; right side plots are for neutron diffraction.