Detecting and Measuring Selection from Gene Frequency Data
55
0
0
Full text
(2)
(3)
(5)
(6)
(7)
(8)
(9)
(10)
(11)
Figure
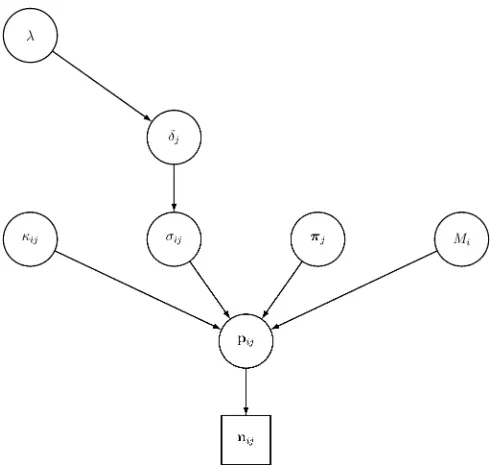
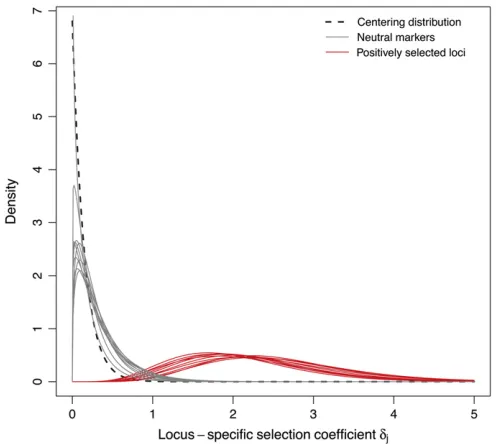
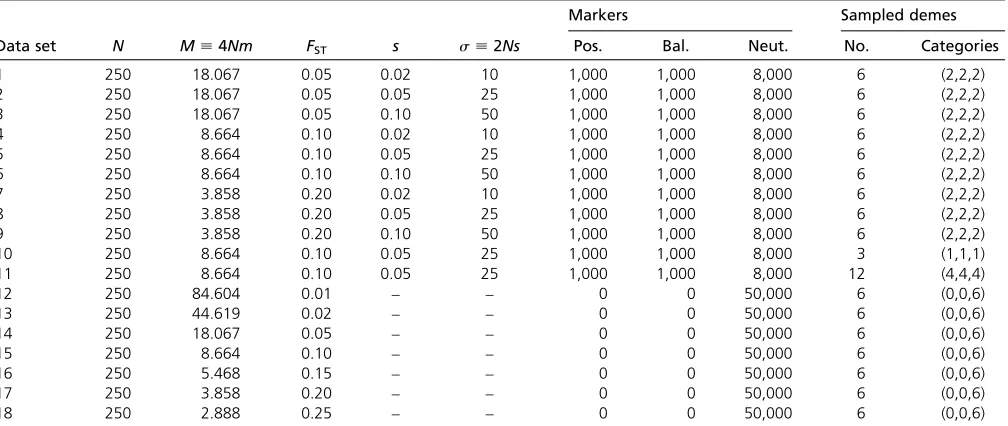
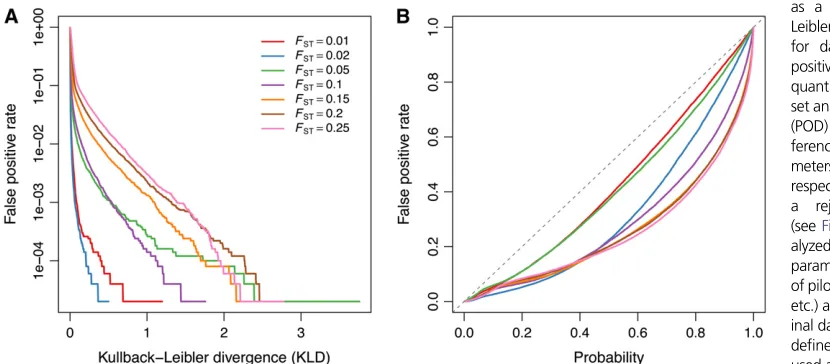
+7

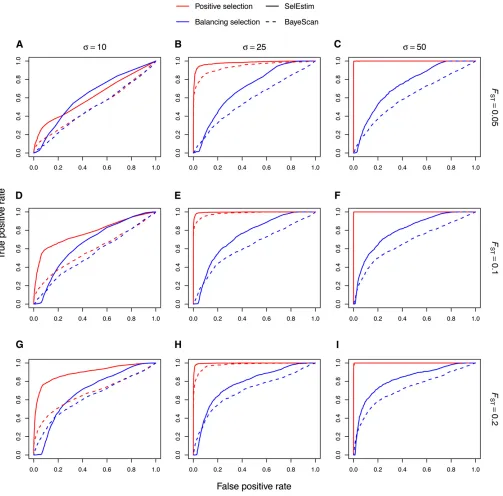
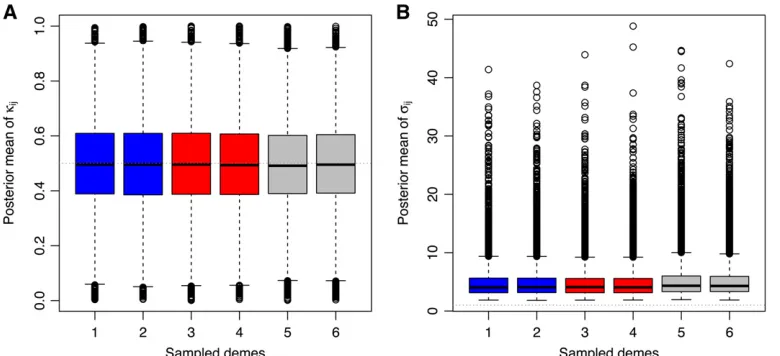
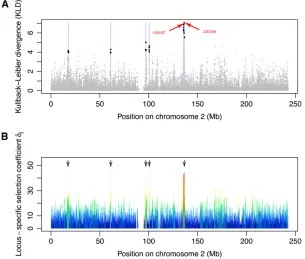

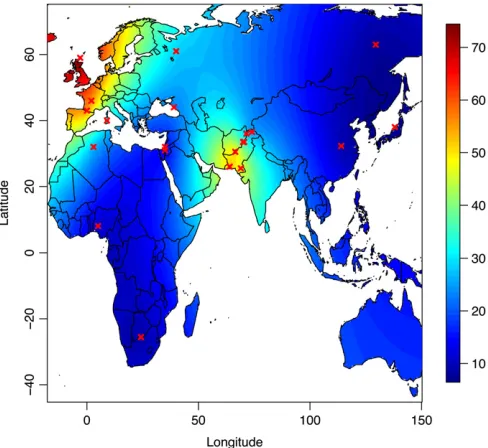
Related documents